- The paper demonstrates that raw sequence tokenization limits representation, while context-driven inputs enhance semantic alignment and reasoning.
- It reveals that modality-based encoders can lose fine-grained mutation sensitivity during alignment, degrading performance on novel protein tasks.
- Empirical benchmarks confirm that context-only inputs outperform sequence-based methods in accuracy, generalization, and computational efficiency.
Resolving the Tokenization Dilemma in Scientific LLMs: The Role of Context in Biomolecular Understanding
Introduction
Accurate interpretation of biomolecular sequences is central to advances in computational biology and bioinformatics. Scientific LLMs (Sci-LLMs) have been pitched as a unified interface between the structure of biological macromolecules and higher-level reasoning. Traditional paradigms treat biomolecular sequences as specialized forms of language or as distinct modalities, but both approaches have severe representational bottlenecks and semantic misalignment issues when interfacing with large-scale LLMs. This paper "Lost in Tokenization: Context as the Key to Unlocking Biomolecular Understanding in Scientific LLMs" (2510.23127) presents a systematic reevaluation of how biomolecular data should be integrated into Sci-LLMs, arguing that the current sequence-centric focus creates more obstacles than solutions for knowledge-based inference.
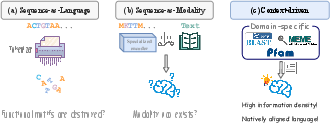
Figure 1: Comparison of integration paradigms—sequence-as-language, sequence-as-modality, and context-driven—for Sci-LLMs.
Tokenization Dilemma: Sequence-as-Language vs Sequence-as-Modality
The "sequence-as-language" approach directly tokenizes biomolecular sequences (proteins, DNA, RNA) into atomic units (amino acids, nucleotides) and feeds them as extended vocabularies into the LLM. This results in weak representations, as tokenization annihilates higher-order information such as motifs or domains essential for biological function. As shown by the t-SNE projections and clustering metrics, sequence-level tokenization yields latent spaces with poor class separability, confirmed by low ARI values (e.g., NatureLM: 0.49; Intern-S1: 0.69).
The "sequence-as-modality" strategy uses specialized encoders (e.g., ESM, SaProt) to inject dense, functionally-aware representations into the transformer, which are then projected into the semantic space of the LLM via dedicated alignment modules (e.g., Q-Former). While representation quality is much higher at the encoder stage, alignment to the LLM’s linguistic latent space results in semantic drift: the fine-grained, evolutionarily-constrained signal from the biological encoder is washed out, substantially reducing the utility of such representations for reasoning and generalization.
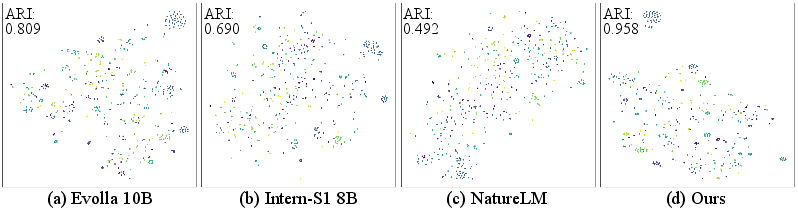
Figure 2: t-SNE visualizations of model representational spaces, demonstrating poor class separation in sequence-as-LLMs, improved structure in modality encoders, and optimal alignment in the context-driven approach.
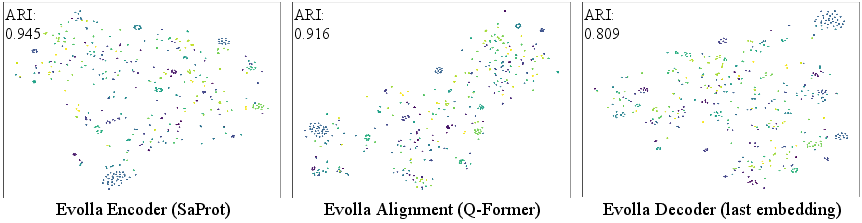
Figure 3: Stepwise degradation of functional structure observed as embeddings progress from the SaProt encoder through the Q-Former alignment and into the LLM in Evolla-10B.
The Context-Driven Paradigm: Empirical Justification
The paper introduces a third, context-driven paradigm. Instead of exposing LLMs to noisy, low-level sequence information, the model is fed high-density, structured natural language context derived from expert bioinformatics tools (e.g., InterProScan, BLASTp, Pfam, ProTrek). This context is natively aligned with the LLM's language domain, allowing the model to leverage its reasoning capacity on rich, human-readable summaries, annotations, and functional predictions.
Benchmarking reveals consistent and substantial superiority of the context-driven approach over both sequence-only or mixed (sequence + context) input modes. Across diverse protein function QA tasks and protein classification datasets, context-only inputs yield the highest mean scores and F1 values, while the addition of raw sequence information often degrades performance rather than providing additive benefit. This is observed both in specialized Sci-LLMs and in general LLMs (e.g., Deepseek-v3, Gemini2.5 Pro, GPT-5).
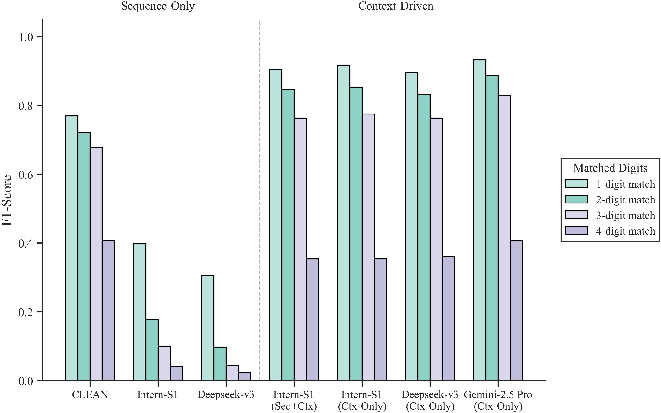
Figure 4: F1 score comparison for EC number prediction shows a robust advantage for context-driven models over specialized sequence-based ones, especially at higher levels of the function hierarchy.
Generalization and Robustness
The context-driven approach demonstrates robust generalization, maintaining high performance on novel protein families (as stratified by sequence identity and publication year) where sequence-centric models fail. Sequence-as-LLMs display flat, baseline-level performance, incapable of extracting meaningful information from novel inputs. Sequence-as-modality models like Evolla exhibit a steep, monotonic drop on "hard" sets or recent proteins, indicative of overfitting and reduced extrapolation capability. In contrast, context-driven models—even when homologous information is sparse—gracefully degrade, validating the resilience of knowledge-based context synthesis over direct sequence decoding.
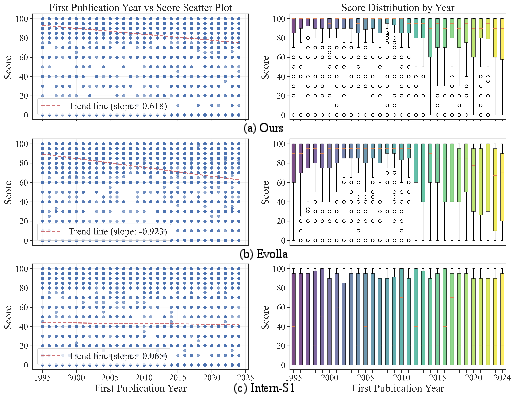
Figure 5: Analysis of model performance versus protein’s first publication year. Context-driven models degrade much less on recently published, "novel" proteins.
Architectural Implications: Representation versus Alignment
The trade-off between the fidelity of biological representation and semantic alignment with language is quantitatively mapped. Sequence-as-LLMs offer maximum alignment but weak representation; modality-based approaches offer richer biological signal but suffer critical alignment loss. Context-driven models leverage the highest possible alignment and representation, with minimal computational cost, decisively dominating the Pareto front.
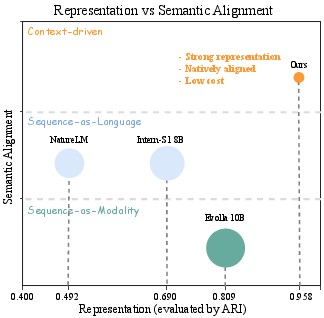
Figure 6: Representation vs. semantic alignment landscape and computational cost tradeoff among sequence, modality, and context-based paradigms.
Biological Reasoning, Semantics, and Sensitivity
Layer-wise feature analyses highlight that Q-Former-style alignments in modern modality-based Sci-LLMs can erase subtle mutation effects, rendering these models ill-suited for fine-grained applications such as variant effect prediction or protein engineering. In contrast, the context-driven approach leverages summary statistics (GO terms, domain compositions), which are naturally robust to noise and highly interpretable, while current context toolchains remain limited in mutation sensitivity due to reliance on homology and motif matching tools.
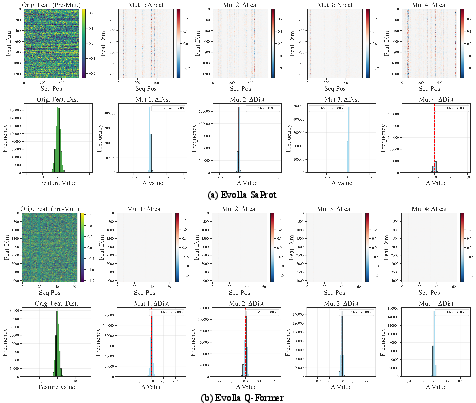
Figure 7: Mutation effect visualization—biological encoders are sensitive to perturbations, but alignment modules erase this signal, diminishing mutation interpretability.
Generalizability Beyond Protein Domains
The context-driven paradigm’s efficacy is demonstrated beyond protein classification, including enzyme function prediction, pathway association, subcellular location inference, and DNA-based disease prediction. Using a standard KEGG DNA pathogenicity prediction task, context-only models again consistently outperform sequence-based approaches, with the addition of sequence serving only to inject noise and reduce output reliability.
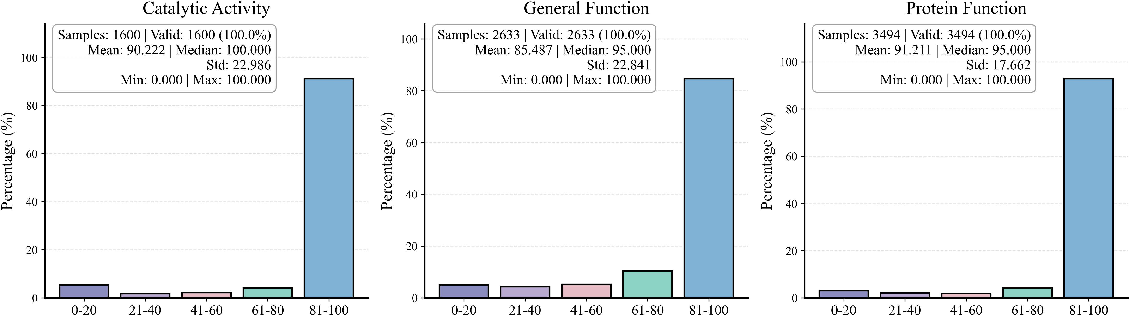
Figure 8: Mol-Instruction protein classification—high mean scores and robust generalization for context-driven approaches across functionally diverse protein tasks.
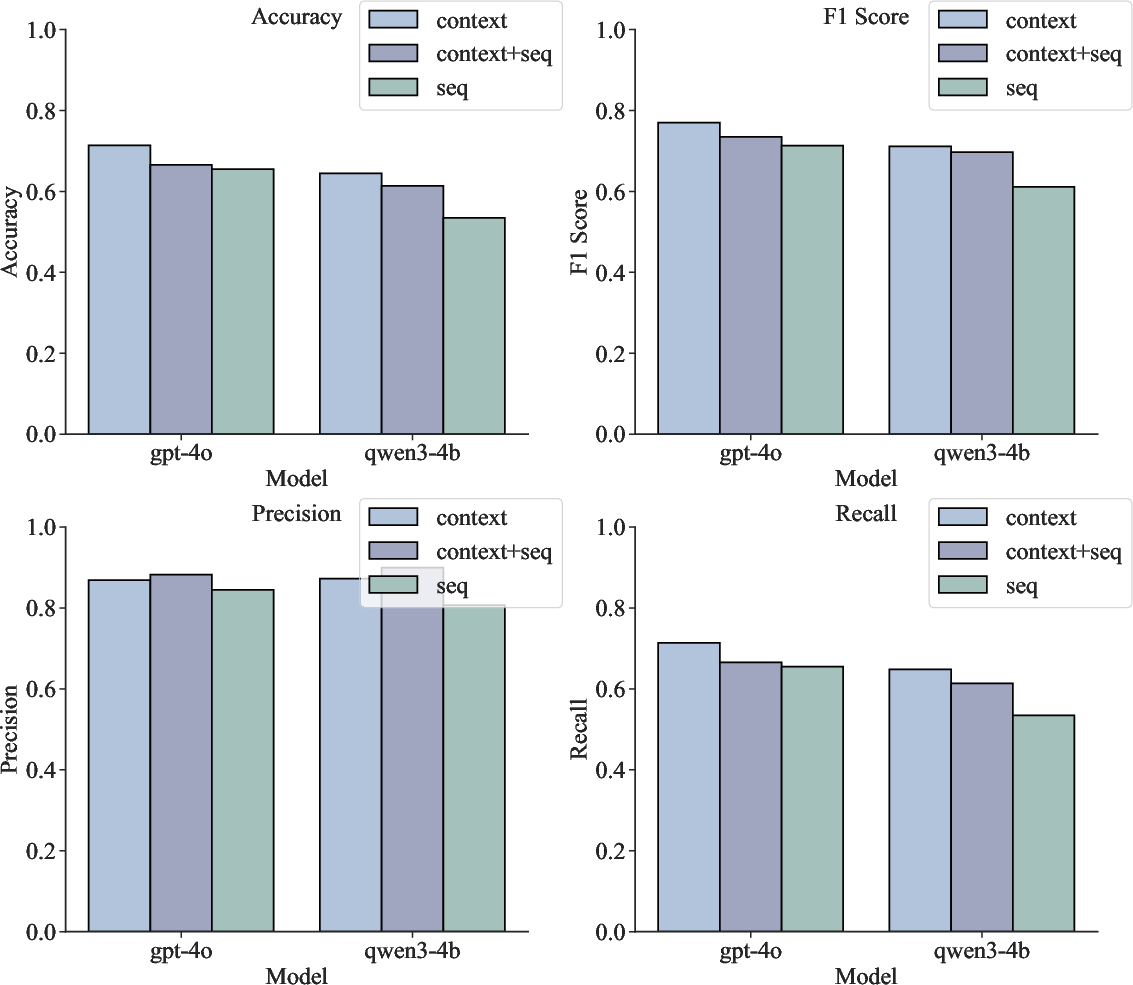
Figure 9: DNA mutation reasoning on KEGG dataset—context-based LLMs outperform or match sequence-based approaches on all metrics.
Implications, Limitations, and Future Directions
This study demonstrates, with robust empirical evidence, that existing Sci-LLMs are not effective de novo sequence interpreters—any observed reasoning power is overwhelmingly due to contextualized expert knowledge, not intrinsic capacity to decode sequence syntax. The research thereby advocates for a paradigm shift: treat LLMs as reasoning engines for high-level knowledge synthesis, not as biological sequence decoders.
Practically, this calls for developing LLM orchestration frameworks that prioritize structured, tool-based evidence (via agents or plug-in tools) and minimize reliance on raw sequence interpretation except where dedicated, highly aligned encoders with proven mutation sensitivity are available. Theoretically, this reframing suggests that the boundary of AI-powered biological discovery will be set not by scale but by advances in extracting, curating, and aligning human-interpretable context for scientific reasoning. Future model designs must address the loss of mutation sensitivity in current context pipelines and explore differentiated architecture designs that can synthesize both fine-grained biochemical signal and high-density expert context within harmonized latent spaces.
Conclusion
The context-driven paradigm provides decisive, validated improvements in accuracy, generalization, and robustness for scientific language modeling in biology, fundamentally shifting the focus of model development from sequence interpretation to high-level knowledge synthesis. Remaining limitations lie in the precision of mutation effect prediction and the comprehensive treatment of non-protein biomolecules. Future AI agents in biology must integrate evolving, context-rich knowledge bases and develop new architectures or toolchains for effective fine-structure and variant-sensitive interpretation.
Figure 6: The context-driven paradigm resolves the tokenization dilemma, achieving the optimal balance between biological representation, semantic alignment, and computational efficiency.

