Reverse Distillation: Consistently Scaling Protein Language Model Representations
Abstract: Unlike the predictable scaling laws in natural language processing and computer vision, protein LLMs (PLMs) scale poorly: for many tasks, models within the same family plateau or even decrease in performance, with mid-sized models often outperforming the largest in the family. We introduce Reverse Distillation, a principled framework that decomposes large PLM representations into orthogonal subspaces guided by smaller models of the same family. The resulting embeddings have a nested, Matryoshka-style structure: the first k dimensions of a larger model's embedding are exactly the representation from the smaller model. This ensures that larger reverse-distilled models consistently outperform smaller ones. A motivating intuition is that smaller models, constrained by capacity, preferentially encode broadly-shared protein features. Reverse distillation isolates these shared features and orthogonally extracts additional contributions from larger models, preventing interference between the two. On ProteinGym benchmarks, reverse-distilled ESM-2 variants outperform their respective baselines at the same embedding dimensionality, with the reverse-distilled 15 billion parameter model achieving the strongest performance. Our framework is generalizable to any model family where scaling challenges persist. Code and trained models are available at https://github.com/rohitsinghlab/plm_reverse_distillation.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper looks at protein LLMs—computer programs that read protein sequences (strings of amino acids) and turn them into numbers that capture patterns and meaning. In many areas like text or images, making models bigger usually makes them better. But for proteins, bigger models often don’t help and can even make things worse. The authors introduce a simple way, called Reverse Distillation, to rearrange what these models output so that bigger always means better and the results are easier to reuse.
What questions did the researchers ask?
They asked:
- Why do bigger protein models sometimes perform worse?
- Can we reorganize the information from big models so it builds neatly on top of what smaller models already know?
- Can we make “nested” embeddings (the numeric representations) so that the first part is exactly the smaller model, and extra parts add new, non-overlapping information?
- Will this fix the scaling problem and improve accuracy on real protein tasks?
How did they approach the problem?
Think of a protein model’s output like a playlist with many tracks mixed together. Small models have room for only the most common and useful tracks (general protein features). Big models add lots of extra tracks (rare or specialized features). When all the tracks are mixed, simple tools can struggle to find the ones that matter for a given task.
Reverse Distillation separates these tracks so they don’t interfere. Here’s the idea in everyday terms:
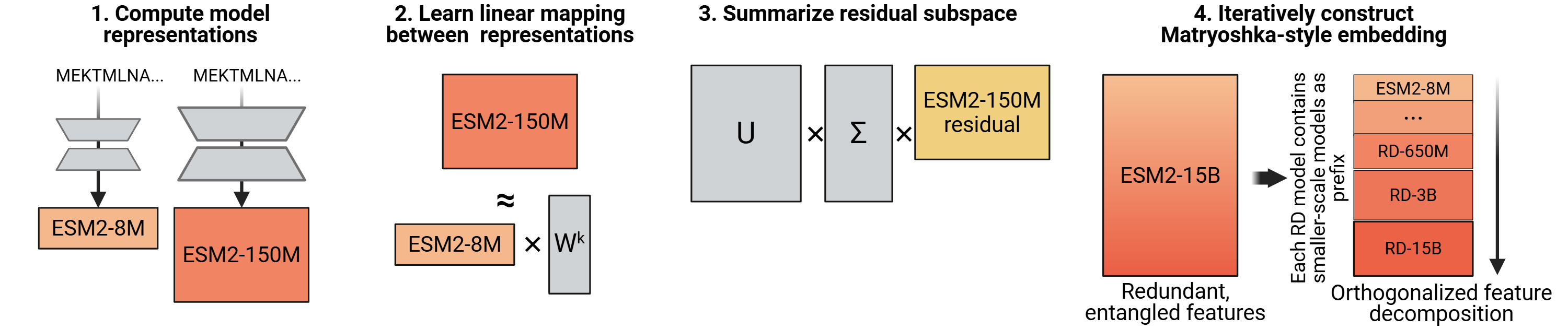
- Start with two models from the same family: a smaller one and a larger one. Both produce a list of numbers for each protein—the “embedding.”
- Keep the smaller model’s embedding exactly as-is. This is your solid foundation (the common, broadly useful features).
- Ask: “Which parts of the big model’s embedding can the small model already predict?” Subtract those out. What’s left are the “leftover” parts unique to the big model.
- Use a math tool (SVD—imagine rotating and lining up a cloud of points so the longest directions come first) to find the most important directions in these leftovers.
- Put the small model’s numbers first, then append the cleaned, non-overlapping “extra” numbers from the big model.
This creates a Matryoshka, or Russian nesting doll, structure: the first chunk of numbers is exactly the smaller model; the next chunk adds the big model’s unique information. If you need a shorter embedding, you can just take the front part. If you need more detail, you include more.
A few helpful definitions:
- Protein LLM (PLM): a model trained on lots of protein sequences to learn patterns, like grammar for proteins.
- Embedding: a list of numbers that summarizes a protein’s features.
- Orthogonal: “non-overlapping” or independent information.
- SVD (singular value decomposition): a way to pick out the most important directions in data, like finding the main axes of a blob so you can describe it efficiently.
They also “chain” this process across many sizes (tiny → small → medium → big → biggest), so each size adds its own clean, non-overlapping chunk of information.
What did they find and why does it matter?
In short: Reverse Distillation makes bigger protein models reliably better and easier to use.
Key results:
- Consistent scaling restored: After reverse distillation, larger models almost always beat smaller ones. This fixes the surprising “bigger is worse” behavior often seen in protein models.
- Better accuracy at the same size: For the same embedding size, reverse-distilled versions usually outperform the original models.
- Top performance: On the ProteinGym benchmark (which measures how mutations affect protein function), the reverse-distilled 15B model achieved the strongest overall results. They also saw gains on other tasks like predicting secondary structure and metal ion binding.
- Nesting works in practice: Because the embeddings are built like nesting dolls, you can use just the first part when you need to save compute or storage, and add more parts if you need more accuracy.
- Reasonable speed: Even though reverse distillation can call several models, the extra time was modest because the smaller models are fast.
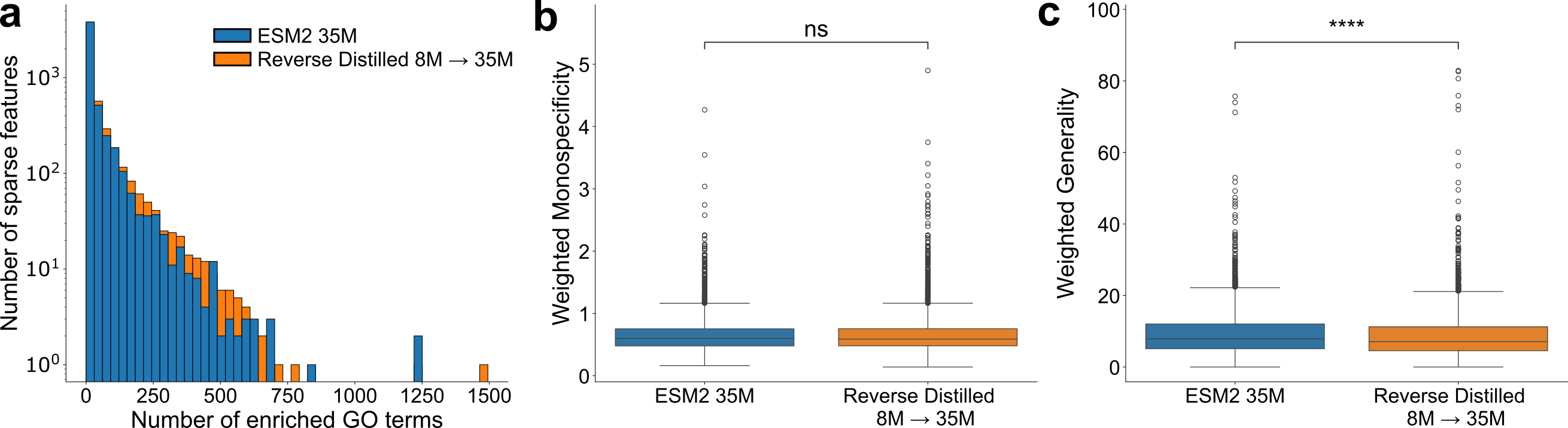
They also tested how well the reorganized embeddings separate biological functions using sparse autoencoders (tools that try to discover meaningful features). The reverse-distilled embeddings showed more function-related signals and less “generic” mixing, suggesting the features are cleaner and easier to interpret.
What does this mean for the future?
- More reliable protein modeling: You can trust that upgrading to a bigger model will help, instead of gambling on whether it might hurt.
- Reusable embeddings: “Embed once, reuse prefixes as needed.” This can save time and storage in pipelines.
- Broad applicability: The same idea could help other kinds of biological models—and even models outside biology—whenever bigger doesn’t automatically mean better.
- Room for improvement: The current method uses simple, transparent math. The authors suggest future work could use nonlinear tools or small fine-tuning tricks to squeeze out even more performance, possibly producing these clean embeddings with just one model pass.
Overall, Reverse Distillation shows that the useful information is already inside big protein models. The trick is organizing it so the basics come first and the extras don’t drown them out—much like building with clear, stackable layers instead of a tangled heap.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a concise, actionable list of what remains uncertain or unexplored in this work and where future research could extend or stress-test the proposed framework.
- Generalization beyond ESM-2: Does reverse distillation (RD) transfer to other PLM families and architectures (e.g., autoregressive models like ProGen, joint sequence–structure models, diffusion models, multimodal protein models), especially when tokenization/positional encodings differ or when embedding dimensionality is not monotonic across scales?
- Nonlinear reverse distillation: How do nonlinear mappings (e.g., kernel ridge, deep regressors, CCA/Deep CCA, contrastive alignment) affect downstream task performance and scaling monotonicity, beyond improved reconstruction noted in the paper?
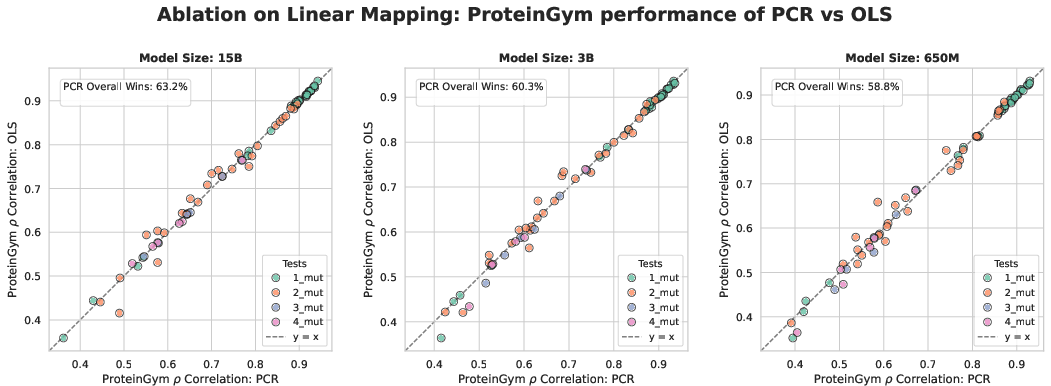
- Theoretical guarantees with PCR: The MSE-optimality theorem is stated for OLS, but training uses principal component regression with noise-PC removal. Do analogous optimality or error bounds hold under PCR, and how do they change with different PC thresholds?
- Residual rank selection: The method fixes residual dimensionality to . Is this always optimal? Can principled rank selection (e.g., via information criteria, cross-validation, random matrix thresholds) improve signal-to-noise and task performance?
- Chaining stability and orthogonality: How do errors and subspace drift accumulate across multi-step chains (e.g., 8M→…→15B)? Are later residuals still (approximately) orthogonal to earlier subspaces in held-out data, and does enforcing explicit orthonormalization across steps help?
- Dataset dependence of the learned subspaces: How sensitive are and to the size, taxonomic diversity, and selection of the 1,000-sequence training set? Would domain-targeted or family-balanced training sets yield better decompositions?
- Robustness to distribution shift: Do RD representations retain their advantages on out-of-distribution proteins (e.g., remote homologs, novel folds, disordered proteins, membrane proteins, metagenomic sequences)?
- Broader task coverage: Effects are shown primarily on ProteinGym DMS and a few property prediction tasks. How does RD impact:
- zero-shot settings (no probe training),
- structure/contact recovery, binding-site prediction,
- protein–protein and protein–ligand interaction tasks,
- stability/thermostability prediction, enzyme specificity,
- long-range dynamics or flexibility (e.g., ATLAS, NMR dynamics)?
- Monotonicity guarantees for downstream tasks: The method is MSE-optimal in a constrained sense but offers no formal guarantee of monotonic downstream performance. Under what assumptions (e.g., task linear separability, noise models) can monotonic improvements be guaranteed?
- Statistical significance and effect sizes: Several improvements are modest and sometimes non-uniform across datasets. Rigorous statistical testing (paired tests, CIs, multiple-comparison correction) and effect-size reporting are needed to substantiate “nearly always scales.”
- Epistasis capture: DMS evaluation averages token-difference vectors for multiple mutations, potentially underrepresenting non-additive effects. Do RD residuals better encode higher-order epistasis than baselines when evaluated with explicit non-additivity metrics or combinatorial mutagenesis datasets?
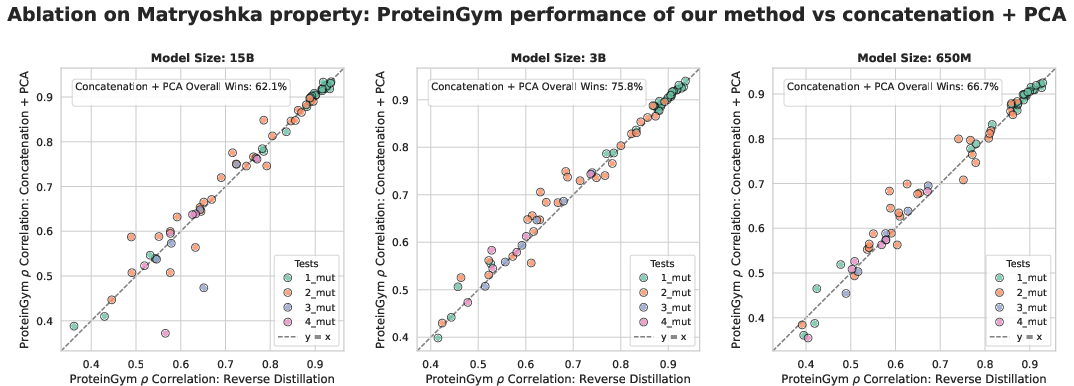
- Matryoshka prefix performance curves: The paper asserts prefix usability but does not quantify performance as a function of prefix length. What are the compute–accuracy trade-offs across prefixes, and do they degrade smoothly across many tasks?
- Alternative baselines: How does RD compare to strong alignment/merging methods such as CCA/PLS, multi-scale feature concatenation with supervised dimensionality reduction, Procrustes alignment, Matryoshka-specific fine-tuning (e.g., Kusupati et al.), or layer-wise alignment rather than final-layer-only?
- Single-pass deployment: RD currently requires running multiple models at inference. Can parameter-efficient fine-tuning (LoRA/adapters) or architectural modifications produce RD embeddings in a single forward pass without sacrificing performance?
- Use cases with partial model access: If only a large model (or only a small model) is available (e.g., API access), can approximate residuals or prefixes be recovered without full-chain inference? What are the accuracy penalties and practical recipes?
- Token-level sampling assumptions: Stacking tokens as samples could bias subspace learning by sequence length distribution and positional effects. How do normalization choices (e.g., per-protein weighting, length stratification) affect and downstream performance?
- Hyperparameter sensitivity: How sensitive are results to PCR thresholds (e.g., Johnstone cutoff), ridge probe regularization, SVD truncation, and sequence filtering criteria? Provide ablations and guidelines for robust defaults.
- Biological interpretability of residuals: Do RD residuals concretely enrich for family-specific motifs, long-range couplings, allosteric signals, or contact patterns? Validate with contact-map analysis, motif discovery, and structure/function annotations beyond GO-term enrichment.
- Task-specific trade-offs: Some tasks (e.g., LOC, MIB at certain scales) show mixed outcomes. Can task-adaptive residual selection or weighted chaining mitigate regressions and deliver per-task optimal prefixes/subspaces?
- Scalability and throughput: Multi-model inference adds overhead. What are the practical limits for proteome-scale embedding, and can caching or prefix reuse strategies materially reduce cost while preserving gains?
- Cross-family/architecture alignment: When model families differ in tokenization, positional encodings, or layer structures, how should representations be aligned for RD (e.g., learned token maps, positional remapping, layer-wise matching)?
- Layer-wise RD: The method uses final-layer embeddings. Does applying RD across corresponding layers (or learned layer pools) yield better decompositions and performance than final-layer-only variants?
- Reproducibility across random seeds and runs: Provide variance across multiple RD training seeds and probe seeds to quantify stability of the decomposition and downstream gains.
- Noise control in residuals: The residual SVD keeps the top components by default; if residuals are low-rank or noisy, this may admit noise. Can automatic noise-floor estimation or shrinkage improve residual quality?
- Generative behavior: RD is evaluated for embeddings and linear probes. How does RD impact generative metrics (e.g., perplexity, likelihood-based probing, sample realism) and does integrating RD into the model’s last layer preserve generation quality?
Practical Applications
Immediate Applications
Below are practical uses that can be deployed now using the paper’s released code/models and simple linear probes, with links to sectors, possible tools/workflows, and feasibility notes.
- Variant effect prediction upgrades for DMS and mutational scanning — sectors: healthcare, biotech, CROs, academia
- Tools/workflows: replace baseline ESM-2 embeddings with reverse-distilled (rd) embeddings in existing ridge/logistic regression pipelines for ProteinGym-like scoring; offer CRO reports that benchmark rd.650/rd.3B/rd.15B alongside baselines.
- Assumptions/dependencies: improvements demonstrated with linear probes on DMS; requires access to the ESM-2 family and the rd decomposition matrices; generalization is best when the target task resembles ProteinGym settings.
- Protein property prediction improvements (SSP-Q3/Q8, metal binding, localization, NMR dynamics proxies) — sectors: biotech, healthcare diagnostics R&D, academia
- Tools/workflows: drop-in rd embeddings for downstream property predictors to boost AU-PR; update internal benchmark suites to rd.15B as default for maximum accuracy.
- Assumptions/dependencies: observed gains are task- and dataset-dependent; downstream heads may need re-tuning; licensing and compute access for ESM-2 models.
- Directed evolution and library design triage — sectors: industrial enzymes, biomanufacturing, therapeutics R&D
- Tools/workflows: use rd embeddings to rank/screen variants pre-experiment; integrate into active-learning loops to prioritize rounds with higher expected fitness.
- Assumptions/dependencies: rd gains measured for linear readouts; lab outcomes still require empirical validation; performance depends on closeness of training distribution to target protein family.
- “Embed once, serve many” with Matryoshka-style prefixes — sectors: software/ML platforms, MLOps for bioAI, cloud compute vendors
- Tools/workflows: centralized embedding store that retains rd.15B vectors and serves k-prefixes (e.g., 1280, 2560) on demand for cost/latency trade-offs; implement API flags like
embedding_dim=1280|2560|5120. - Assumptions/dependencies: storage budget for full embeddings; consistent versioning of rd chains; users accept slight quality differences across prefixes.
- Tools/workflows: centralized embedding store that retains rd.15B vectors and serves k-prefixes (e.g., 1280, 2560) on demand for cost/latency trade-offs; implement API flags like
- Adaptive compute routing for large-scale proteome annotation — sectors: bioinformatics platforms, data providers (e.g., UniProt, Ensembl), national labs
- Tools/workflows: run small-prefix queries by default (fast/cheap) and escalate to longer prefixes for challenging sequences; cache per-protein prefixes for repeated tasks.
- Assumptions/dependencies: batch scheduling that mixes multiple ESM scales; modest 1.5–1.7× inference overhead vs. a single largest model is acceptable.
- Reverse-distilled feature interpretability with sparse autoencoders (SAEs) — sectors: academia, interpretability/ML research, biotech hypothesis generation
- Tools/workflows: train SAEs on rd embeddings to extract more functionally enriched GO features; use for target deconvolution and functional hypothesis generation.
- Assumptions/dependencies: enrichment quality depends on annotation depth; interpretability aids hypothesis generation, not definitive mechanism discovery.
- Robust model selection policy for PLM-based pipelines — sectors: biotech, CROs, ML platform teams
- Tools/workflows: adopt a simple policy “use rd.15B (else rd.3B) by default” to restore monotonic scaling and reduce ad hoc per-task model size tuning.
- Assumptions/dependencies: relies on availability of the model family and the rd chain; tasks outside tested regimes may still warrant validation.
- Turnkey plug-ins for popular protein ML stacks — sectors: software/tools (Hugging Face, bioAI SDKs), platform integrators
- Tools/workflows: package rd inference as a Python/CLI plugin that wraps ESM-2 forward passes, PCR, SVD components; add optional ridge probe training helper for common tasks.
- Assumptions/dependencies: users have GPUs or CPU clusters for batch inference; maintainers keep rd mappings synced to model versions.
- Enhanced QA for PLM scaling studies — sectors: academia, benchmark consortia (e.g., ProteinGym), journals
- Tools/workflows: include rd vs. baseline comparisons in benchmark leaderboards; report scaling monotonicity metrics alongside accuracy.
- Assumptions/dependencies: community adoption; standardization of evaluation scripts and seeds.
- Cost-aware screening in early R&D — sectors: startups/SMEs in biotech, non-profits
- Tools/workflows: run rd.650 or rd.3B prefixes in early triage to cut GPU costs, then escalate to rd.15B for final shortlists; integrate into LIMS/ELNs as a structured step.
- Assumptions/dependencies: approximate monotonic gains hold; acceptable trade-offs between cost and accuracy for early phases.
- Data repository add-ons (pre-computed embeddings) — sectors: data providers, community portals
- Tools/workflows: host pre-computed rd embeddings for reference proteomes; provide download bundles by prefix length and species.
- Assumptions/dependencies: storage and bandwidth budgets; licensing for redistribution; versioning and provenance tracking.
- CRO services for multi-mutation modeling — sectors: CROs, pharma partners
- Tools/workflows: offer reports that leverage rd gains noted for multi-mutant datasets (e.g., averaging per-position deltas) with clearer confidence intervals.
- Assumptions/dependencies: multi-mutation effects are still hard; linear aggregation is a simplification; communicate uncertainty appropriately.
Long-Term Applications
These opportunities likely need further research, scaling, validation, or productization before broad deployment.
- Single-pass reverse-distilled heads via parameter-efficient fine-tuning — sectors: software/ML tooling, platform providers
- Tools/products: LoRA/adapter-tuned large models that output rd embeddings directly in one forward pass; drop the multi-model overhead.
- Assumptions/dependencies: requires successful training to preserve Matryoshka property; engineering for stability and inference speed.
- Nonlinear reverse distillation for stronger residual capture — sectors: academia, advanced ML for bio, platform providers
- Tools/workflows: kernelized or neural predictors for the small→large mapping plus nonlinear residual factorization; potentially higher downstream lift than linear PCR+SVD.
- Assumptions/dependencies: more complex training; risk of reduced interpretability; need to retain nested-prefix guarantees.
- Generative design control via scale-separated features — sectors: protein design, therapeutics, industrial enzymes
- Tools/products: conditioning/guidance interfaces that separately modulate “broad” vs. “specific” features (shared by small vs. unique to large) to steer design objectives.
- Assumptions/dependencies: requires adapting rd to generative model internals; extensive wet-lab validation for efficacy and safety.
- Clinical variant interpretation as supportive computational evidence — sectors: healthcare, clinical genomics, regulators
- Tools/workflows: use rd-boosted predictors as one line of ACMG-supporting evidence for VUS triage and case prioritization.
- Assumptions/dependencies: prospective clinical validation and calibration; regulatory acceptance; clear guardrails that these are supportive, not definitive diagnostics.
- Cross-domain generalization to other bio foundation models — sectors: genomics (DNA/RNA LMs), chemistry (molecular LMs), single-cell, imaging
- Tools/workflows: apply rd chaining to model families with poor scaling; publish “rd packs” for each domain with pre-trained mappings.
- Assumptions/dependencies: availability of size-tiered model families; confirm that small models encode broadly shared features in each domain.
- Standardization of Matryoshka embedding formats and repositories — sectors: data infrastructure, standards bodies, open-source consortia
- Tools/products: a community spec for nested embeddings (prefix alignment, metadata), plus registries that host versioned rd embeddings for common datasets.
- Assumptions/dependencies: community consensus; stewardship for long-term maintenance; legal/licensing clarity.
- Hardware/runtime acceleration of rd pipelines — sectors: GPU vendors, inference providers, compiler/toolchain teams
- Tools/products: fused kernels to run multiple model scales efficiently; caching/streaming of prefixes; distributed inference graphs optimized for rd chaining.
- Assumptions/dependencies: engineering investment; demand at scale; co-design with framework maintainers.
- Epistasis-aware active learning loops — sectors: protein engineering, directed evolution
- Tools/workflows: use rd residual subspaces to better detect non-additive interactions; guide combinatorial library construction and experimental design.
- Assumptions/dependencies: requires method development to map residual components to epistatic patterns; iterative wet-lab feedback.
- Policy and funding guidance for efficient scaling of bio foundation models — sectors: policy, funders, public-private partnerships
- Tools/workflows: evaluation protocols that include scaling monotonicity and efficiency metrics; funding calls that prioritize methods improving reuse (e.g., prefix embeddings).
- Assumptions/dependencies: stakeholder engagement; empirical evidence of cost/benefit in public datasets and national infrastructures.
- Educational/benchmark platforms for multi-scale representation analysis — sectors: education, community benchmarks
- Tools/products: interactive labs illustrating bias-variance tradeoffs and rd decomposition; benchmark tracks assessing interpretability and feature disentanglement.
- Assumptions/dependencies: curated curricula and maintainers; ongoing updates as models evolve.
- Safety and governance overlays for accelerated protein design — sectors: policy, biosecurity, ethics
- Tools/workflows: risk assessment checklists and model-use monitoring when rd-enabled pipelines increase design throughput; red-teaming benchmarks.
- Assumptions/dependencies: coordination with domain experts and regulators; integration with broader responsible AI frameworks.
Notes on general feasibility
- Dependencies: access to the ESM-2 model family, GPUs/accelerators, and the provided rd code/models; small training corpus (∼1k sequences) suffices for linear mappings in the shown setup.
- Scope of gains: clearest benefits are demonstrated with linear probes on ProteinGym-style tasks and several property prediction datasets; other tasks (e.g., structure prediction, generative use) require validation.
- Operational overhead: current rd inference invokes multiple model scales but adds only ∼1.5–1.7× latency relative to a single largest model; single-pass variants are a promising future optimization.
- Generalization risk: improvements rely on the assumption that smaller models encode broadly shared features and that larger models’ unique features can be cleanly isolated; this should be re-checked per new model family/domain.
Glossary
- Adaptive SVD: A technique that adapts singular value decomposition for continual or constrained learning settings to maintain subspace separation. "o-LoRA~\citep{wang2023orthogonal} and Adaptive SVD~\citep{nayak2025sculpting}, which maintain orthogonal weight subspaces when adapting to new tasks."
- Amino acid alphabet: The set of standard amino acid symbols used to represent protein sequences. "where is the amino acid alphabet,"
- Allosteric couplings: Long-range interactions in proteins where binding at one site affects activity at another. "protein family-specific allosteric couplings,"
- Allosteric signals: Signals in proteins indicating allosteric regulation, where distant sites influence each other’s function. "allosteric signals."
- Bias–variance tradeoff: The balance between model complexity (variance) and simplicity (bias) that affects generalization. "A motivating intuition for our approach comes from the bias-variance tradeoff."
- Contact-recovery signals: Signals indicating how well a model recovers residue–residue contacts from sequence. "delivers contact-recovery signals comparable to the 15B variant"
- Cylinder set: In this context, the set of higher-dimensional representations that preserve a lower-dimensional prefix. "the cylinder set of in "
- DAG (Directed Acyclic Graph): A graph with directed edges and no cycles; used to structure Gene Ontology terms. "the DAG relationship between GO terms"
- Deep mutational scanning (DMS): High-throughput experiments measuring effects of many mutations on protein function. "on deep mutational scanning (DMS) benchmarks from ProteinGym"
- Eckart–Young theorem: A result stating that truncated SVD gives the best low-rank approximation in least squares sense. "The proof directly follows from the Eckart-Young theorem."
- Epistatic interactions: Non-additive interactions between mutations where the combined effect differs from the sum of individual effects. "epistatic interactions,"
- Gene Ontology (GO) terms: Controlled vocabulary terms describing gene and protein functions, processes, and locations. "identify the set of GO terms associated with each sparse feature."
- Generality: A metric indicating how high-level or broad the GO terms associated with a feature are. "and \"generality,\" the depth of the least common ancestor (LCA) of each pair of terms in the set"
- Hydrophobicity patterns: Patterns in proteins related to hydrophobic amino acid distribution, influencing folding and function. "hydrophobicity patterns,"
- Johnstone threshold: A criterion from random matrix theory used to distinguish signal from noise in principal components. "apply the Johnstone threshold~\citep{johnstone2001distribution} from random matrix theory"
- Least common ancestor (LCA): The deepest shared ancestor of two nodes in a hierarchy, used here on the GO DAG. "the depth of the least common ancestor (LCA) of each pair of terms in the set"
- Leave-one-out cross-validation: A validation method where each sample is used once as the test set while the rest form the training set. "We fit the ridge regression on 80\% of the single-mutational variants using leave-one-out cross-validation."
- Linear probes: Simple linear models trained on fixed embeddings to evaluate the information content of representations. "linear probes on larger, mixed representations frequently fail to isolate task-relevant signal."
- LoRA (Low-Rank Adaptation): A parameter-efficient fine-tuning method that injects low-rank updates into pretrained weights. "parameter-efficient fine-tuning (e.g., LoRA)"
- Manifold: A mathematical space that locally resembles Euclidean space; here, the set of embeddings produced by a model. "Let be the manifold spanned by embeddings of ."
- Matryoshka-style embeddings: Embeddings designed so that prefixes are themselves valid, progressively improving representations. "yields Matryoshka-style embeddings where each prefix corresponds to a valid reverse-distilled representation at that scale."
- Monospecificity: A metric measuring how tightly a feature’s associated GO terms cluster in the ontology. "we compute \"monospecificity,\" the inverse of average shortest pairwise distance between GO terms in the same set"
- Multiple sequence alignment (MSA): An alignment of three or more sequences to reveal conserved patterns and relationships. "hybrids leveraging MSAs or structure often winning on zero-shot fitness"
- Orthogonal complement: The subspace consisting of vectors orthogonal to a given subspace; used to isolate new information. "By computing the orthogonal complement of the smaller model's subspace within the larger model's representation,"
- Orthogonal subspaces: Subspaces with zero inner product, enabling decomposition without interference between components. "we decompose the representation space into orthogonal subspaces:"
- Parameter-efficient fine-tuning: Techniques that adapt large models by training a small number of new parameters. "parameter-efficient fine-tuning (e.g., LoRA)"
- Principal component regression (PCR): Regression performed on principal components, often for denoising or multicollinearity control. "we use principal component regression (PCR) rather than ordinary least squares."
- Protein LLM (PLM): A model trained on protein sequences to learn representations capturing biological patterns. "Protein LLMs (PLMs) have emerged as powerful representation learners,"
- ProteinGym: A benchmark suite for protein fitness prediction and related tasks. "On ProteinGym benchmarks,"
- Random matrix theory: A field studying properties of matrices with random elements, used here for component selection. "from random matrix theory"
- Residue co-evolution patterns: Statistical dependencies between amino acid positions that co-vary across sequences. "Each model learns residue co-evolution patterns through self-attention mechanisms,"
- Reverse Distillation: The paper’s framework that uses smaller models to structure and decompose larger models’ representations. "We introduce Reverse Distillation, a principled framework that decomposes large PLM representations into orthogonal subspaces guided by smaller models of the same family."
- Ridge regression: A linear regression method with L2 regularization to mitigate overfitting and multicollinearity. "feeding it into a ridge regression classifier."
- Singular value decomposition (SVD): A matrix factorization method that decomposes a matrix into singular vectors and values. "Apply SVD: "
- Sparse autoencoder (SAE): An autoencoder with sparsity constraints on hidden units to learn interpretable features. "training sparse autoencoders (SAE)."
- Spearman correlation: A rank-based correlation coefficient assessing monotonic relationships between variables. "we computed the Spearman correlation between our predicted scores and the ground truth"
- UMAP (Uniform Manifold Approximation and Projection): A nonlinear dimensionality reduction technique preserving local structure. "Nonlinear dimensionality reduction (e.g., UMAP~\citep{mcinnes2018umap}) could also more effectively separate residual features,"
- UniProt: A comprehensive protein sequence and annotation database. "Using a set of 18,142 proteins from the UniProt database,"
- UniRef50: A clustered protein sequence database at 50% identity to reduce redundancy. "For training, we used sequences sampled randomly from UniRef50;"
- Variant-effect accuracy: Performance measure of predicting functional impact of mutations (variants) in proteins. "variant-effect accuracy peaks at intermediate model perplexity,"
Collections
Sign up for free to add this paper to one or more collections.