- The paper surveys AI methods for protein dynamics by integrating deep generative models with physical energy functions to generate equilibrium ensembles.
- It details techniques such as flow matching, diffusion models, and autoregressive strategies that improve conformational sampling in proteins.
- It highlights challenges in scalability, experimental data integration, and unbiased ensemble estimation with implications for drug discovery.
Artificial Intelligence for Protein Dynamics: Structure, Energy, and Dynamics Integration
Introduction
Characterizing and understanding the dynamic behavior of proteins is central to modern structural biology, mechanistic biochemistry, and rational therapeutic design. The static picture provided by single experimental or predicted structures is fundamentally insufficient for elucidating function, allostery, and binding phenomena, which are inherently multi-state and pathway-dependent. "Learning Structure, Energy, and Dynamics: A Survey of Artificial Intelligence for Protein Dynamics" (2604.25244) provides a comprehensive and technically rigorous review of how deep learning and generative AI have transformed every aspect of modeling protein dynamics, from equilibrium ensemble sampling and surrogate dynamics generation to the direct learning of physically grounded energy landscapes and ML-accelerated MD simulation pipelines.
Protein Dynamics: Motivations, Challenges, and Opportunities
Proteins exhibit high-dimensional, rugged energy landscapes with diverse conformational substates, slow transition kinetics, and long-range allosteric couplings that cannot be adequately probed by finite MD trajectories or structure prediction alone. Landmark phenomena such as functional switching, ligand-induced conformational selection, and disorder-to-order transitions illustrate the essential role of an ensemble- and trajectory-level view over single-structure paradigms.
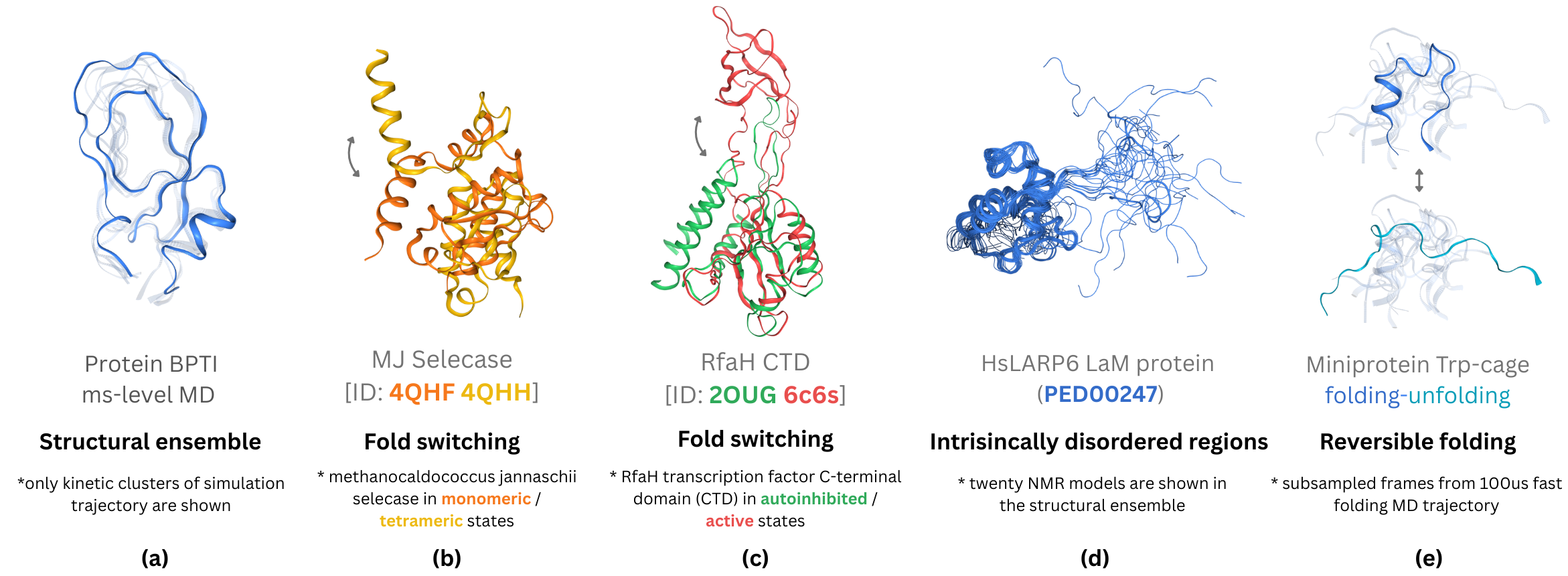
Figure 2: Examples of biomolecular conformational dynamics, including equilibrium ensembles, fold switching, activation states, IDRs, and protein folding snapshots.
However, the computational bottleneck of all-atom MD integration with empirical or quantum force fields, the limitations of empirical FFs for both folded and disordered proteins, and the extreme sparsity of experimental dynamic data underscore the pressing need for data-driven surrogates, generative frameworks, and hybrid energy-structure learning pipelines.
Generative Modeling from Structural Ensembles and Simulation Data
The first major pillar in this survey is the application of generative modeling to static and dynamic protein data. Generative models—normalizing flows, diffusion models, and autoregressive architectures—can learn to sample from p(x∣s), the equilibrium ensemble over structures conditional on sequence, or from p(x1:T∣x1,c), the trajectory distribution for a system initiated from conformation x1 under context c (e.g., temperature, ligand presence).
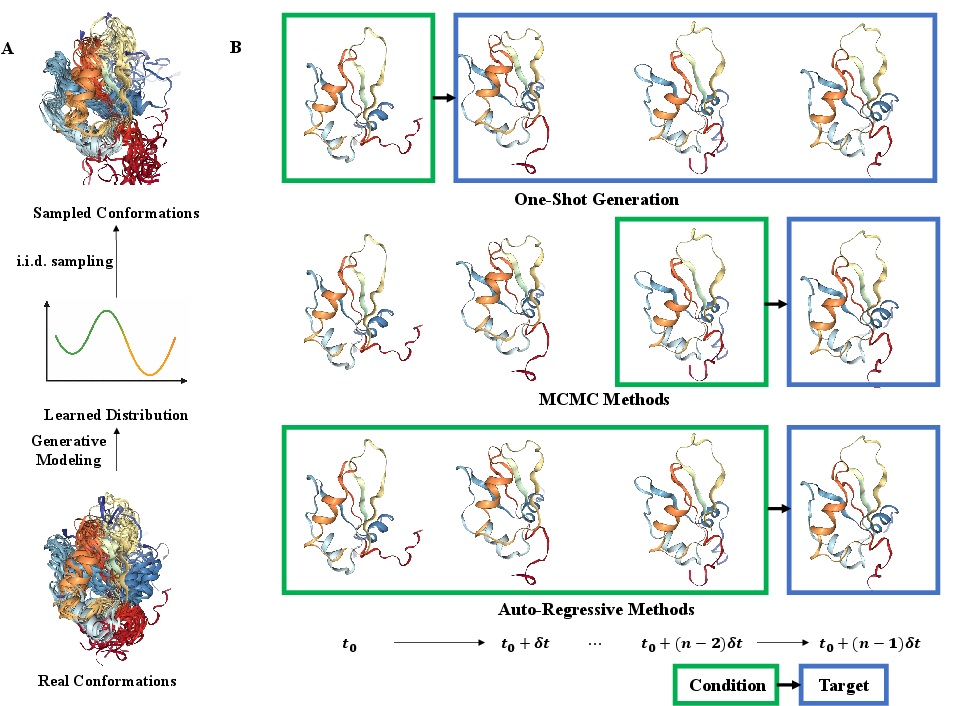
Figure 1: Overview of generative modeling from structure; conformation generation and categories of trajectory generation strategies.
Early successes involved manipulating static predictors (e.g., AlphaFold2) with randomized MSAs but lacked formal statistical guarantees and diversity. Recent paradigms have formalized conformational sampling with explicit generative objectives (e.g., flow matching, denoising diffusion) and direct training on simulation or experimental ensembles. For instance, AlphaFlow and ESMDiff fine-tune leading sequence-to-structure models with flow/diffusion objectives for direct ensemble modeling, enabling physically diverse and MSA-free generation [jing2024alphaflow], [lu2025slm]. ConfDiff incorporates force-based priors for physics-guided sampling, yielding improved agreement with Boltzmann-distributed MD [wang2024confdiff]. The extension to protein-protein and protein-ligand systems is exemplified by DynamicBind and PLACER, which handle complex assembly dynamics and ligand-bound conformational heterogeneity.
Three trajectory generation strategies are highlighted: frame transition models (e.g., TimeWarp, EquiJump, DeepJump), autoregressive models (e.g., ConfRover, STAR-MD, MD-LLM-1), and one-shot video-style generation (e.g., MDGen, BioMD, AlphaFolding) [jing2024mdgen], [shen2025confrover], [cheng2025alphafolding]. Autoregression provides kinetic realism and enables kinetic observables estimation; one-shot methods offer efficiency and flexible supervision via masking paradigms.
Learning from Physically-Grounded Energy Functions
Complementary to structural learning, a growing body of work exploits access to physical energy and force functions (either empirical or quantum mechanical) for direct generation of equilibrium ensembles via sampling from the Boltzmann distribution p(x)∝exp(−βE(x)). This approach is formalized by the Boltzmann generator (BG) paradigm, which pairs an expressive (typically invertible) generative model fθ (e.g., normalizing flows, flow-matched diffusion) with tractable model-likelihood evaluation for importance reweighting and unbiased observable estimation.
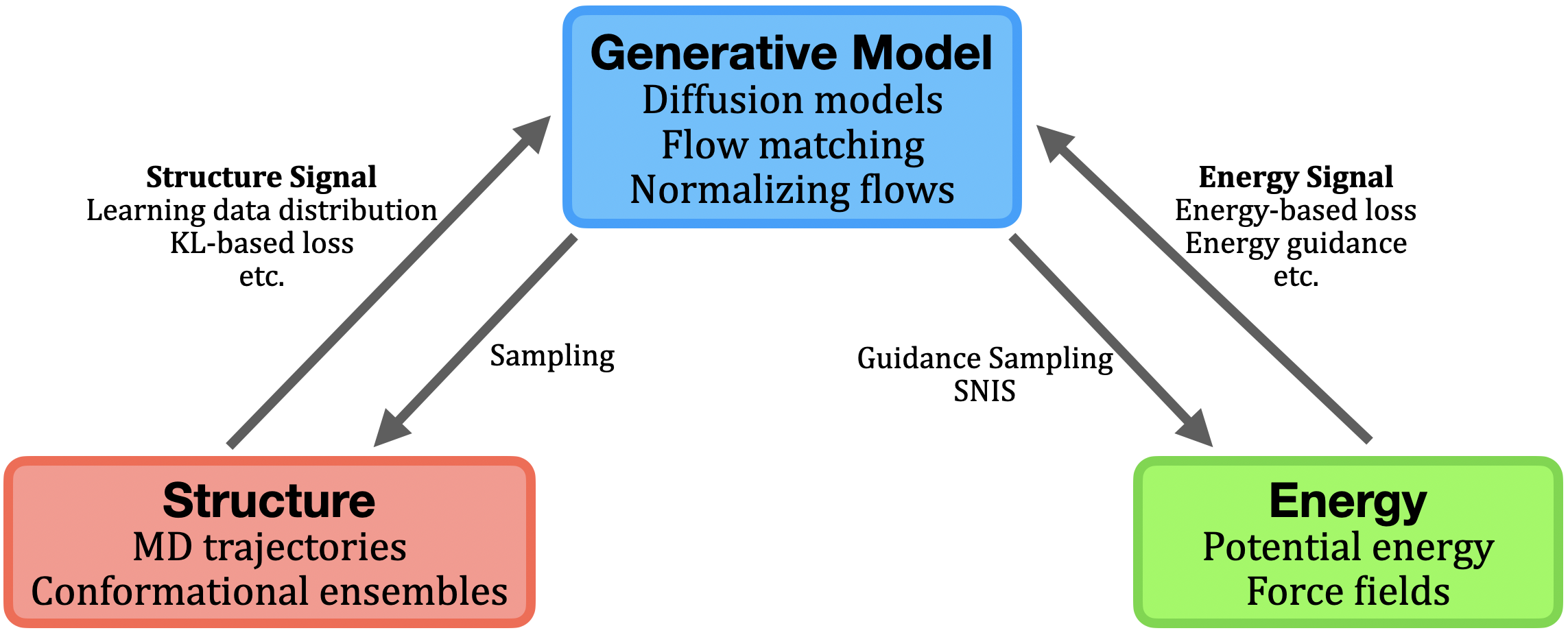
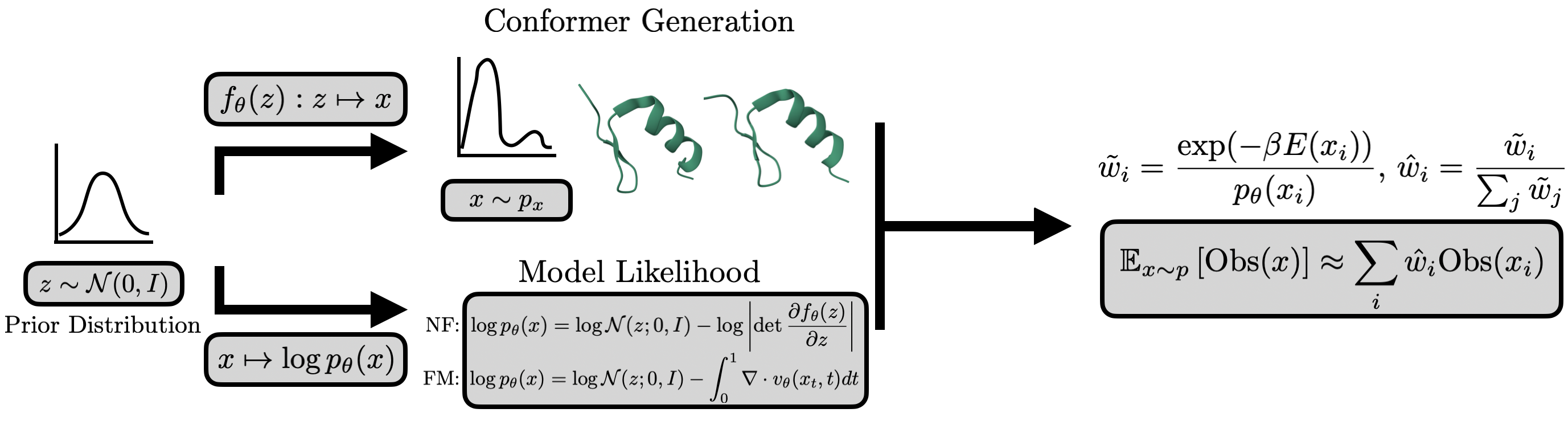
Figure 3: Schematic of structural, energetic, and generative model interactions; BG workflow and SNIS correction for observable recovery.
Architectural and training innovations have enabled BGs to scale from small peptides to larger systems, with transferable (system-conditioned) flows (PROSE), energy-guided amortization, and annealed or importance-weighted corrections to address model-data mismatch and energy landscape ruggedness [noe2019bg], [tan2025prose], [klein2024tbg], [kim2024scalable]. Diffusion-based amortized approaches (iDEM, BNEM, PITA) leverage direct energy/force supervision during generative training, sidestepping the need for equilibrium data. Physics-aware adaptation methods further introduce energy or collective-variable rewards post hoc to steer pretrained structural generators toward more physically faithful distributions or enhanced exploration (EPO, EBA, Metadiffusion) [sun2025epo], [lam2026metadiffusion].
The core technical challenge remains the tension between tractable likelihoods (required for exact reweighting) and scalable, symmetry-aware invertible architectures. Advances in continuous-time flows, flow matching, and hybrid likelihood-free generators represent active progress toward resolving this issue.
Machine Learning for Molecular Dynamics Simulation: Potentials, Coarse-Graining, and CV Discovery
The integration of deep learning into the MD pipeline is driving improvements in both accuracy and efficiency. The fundamental task is the learning of machine-learned potentials (MLPs)—flexible energy function approximators with GNN or hierarchical architectures—from QM or MD data to enable fast, stable, and accurate force computations.
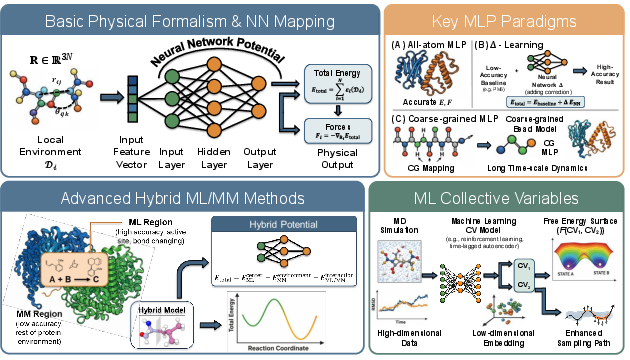
Figure 4: Machine learning potentials: mapping from local environments to total energy and force, hybridization with MM, and ML-driven CV discovery strategies.
GEMS, AI2BMD, Espaloma, and SO3LR illustrate the application of MLPs to large proteins, hybrid QM/MM regions, and explicit solvent environments [unke2024gems], [takaba2024espaloma], [wang2024ai2bmd], [kabylda2025so3lr]. Systematic Δ-learning boosts accuracy by learning quantum corrections. Rollout stability and nonlocal interaction modeling are addressed via explicit long-range terms, global attention, and plug-in diffusion-style modules [hong2026geometric].
For simulation acceleration, ML-based coarse-grained (CG) models—via GNNs, flows, and score-based methods—enable time- and length-scale extension. Pioneering works (CGnets, CGSchNet, TorchMD-Net, ScoreMD) have established generative and force-matching strategies for many-body CG potentials [wang2019cgnet], [charron2025cgschnet], [plainer2025consistent]. Recent methods employ stochastic interpolants, Fokker-Planck regularization, and variational frameworks for physical and kinetic consistency.
Automatic collective variable (CV) discovery, essential for enhanced sampling, benefits from both unsupervised (autoencoder, slow-mode extraction) and RL-based or generative frameworks. Notable advances include DeepTICA for kinetic slow modes, DiffSim for path-based biasing, and BioEmu-CV for time-lagged generative supervision [luigi2021deeptica], [sipka2023diffsim], [parklearning]. ML-based committor estimation (e.g., via GVP-GNNs, Siamese networks, iterative variational schemes) provides interpretable reaction coordinates and transition states [arredondo2025gvpgnn], [megÍas2026ivl].
Data Resources and Systematic Bias
The effectiveness of generative AI in protein dynamics hinges on large, curated datasets: experimental structures (PDB, AFDB), long and diverse MD trajectory repositories (ATLAS, DynamicPDB, mdCATH), and experimental ensemble measurements (BMRB, SASBDB, MobiDB, DisProt). However, static structure databases carry inductive and coverage limitations, while simulation-based datasets reflect force field biases and finite sampling. Emergent approaches include synthetic, distillation-enriched, and hybrid experiment-driven datasets, with meticulous attention to filtering, validation, and integration strategies.
Open Challenges, Implications, and Future Directions
Several fundamental challenges remain at the interface of generative modeling, simulation, and thermodynamic estimation:
- Generative force fields and likelihood-free dynamics: Moving beyond supervised MLP distillation, generative force fields learned via trajectory or structure data alone (without explicit energy labels) are a critical yet open direction. Ensuring physicality, symmetry, and conservative fields is nontrivial.
- Scalable BGs and unbiased ensemble estimation: Scaling proposal models and SNIS corrections to protein complexes with >1000 residues, flexible ligands, and realistic environments remains computationally and algorithmically challenging.
- Integration of experimental constraints: Rigorously fusing sparse, heterogeneous experimental signals (chemical shifts, SAXS, cryo-EM densities) into end-to-end differentiable and generative pipelines is essential for overcoming simulation-induced bias, particularly in IDP or large-scale assembly contexts.
- Rigorous benchmarking and uncertainty quantification: Defining standards for trajectory fidelity, ensemble overlap, and physicality is critical for reproducibility, model selection, and uncertainty-aware simulation rollouts.
- Biomolecular foundation models: The emergence of unified pretraining architectures (e.g., Boltz-2, PROTDYN) that can generalize across diverse systems, tasks, and dynamic phenomena with zero-shot transfer and efficient adaptation is likely to define the next phase of this field [passaro2025boltz], [liu2026protdyn].
Conclusion
This survey achieves a rigorous synthesis of the recent advances in AI for protein dynamics, bridging structural, energetic, and dynamic axes. The ongoing integration of deep generative models with physical supervision, hybrid data sources, and uncertainty-aware inference is poised to not only advance basic biophysical understanding but also accelerate applications in drug discovery, enzyme engineering, and synthetic biology. Open research frontiers remain in the faithful emulation of biological time-scales, the scaling of unbiased generators, and the incorporation of direct experimental feedback, all of which are critical for a comprehensive, predictive model of protein function in motion.