- The paper presents a generative, Euclidean-equivariant flow matching model that generates conformational transitions, accelerating protein MD simulation.
- It demonstrates a systematic trade-off between simulation fidelity and computational speed, achieving up to 1000× acceleration under moderate jump sizes.
- The model replicates essential dynamical properties in fast-folding proteins, offering practical insights for applying deep learning to MD simulations.
Accelerating Protein Molecular Dynamics Simulation with DeepJump
Introduction
DeepJump introduces a generative, Euclidean-equivariant flow matching model for protein molecular dynamics (MD) simulation, targeting the persistent challenge of bridging the gap between atomistic simulation fidelity and biologically relevant timescales. Traditional MD, while physically grounded, is computationally constrained by the need to resolve high-frequency atomic motions, limiting accessible simulation windows to microseconds for most systems. Deep learning-based approaches have made significant progress in static structure prediction and ensemble generation, but generalizable, high-fidelity kinetic modeling of protein dynamics remains underexplored. DeepJump addresses this by learning to generate conformational transitions directly from trajectory data, enabling large, physically plausible jumps in conformational space and thus accelerating the exploration of protein dynamics.
Model Architecture and Training
DeepJump employs a two-stage architecture: a current-step conditioner and a generative transport network, both built on Euclidean-equivariant neural networks. The model is trained on the mdCATH dataset, which provides a diverse set of protein trajectory snapshots across a wide range of folds and temperatures. The training objective is to predict the future 3D state of a protein given its current state, sequence, and a specified jump size δ.
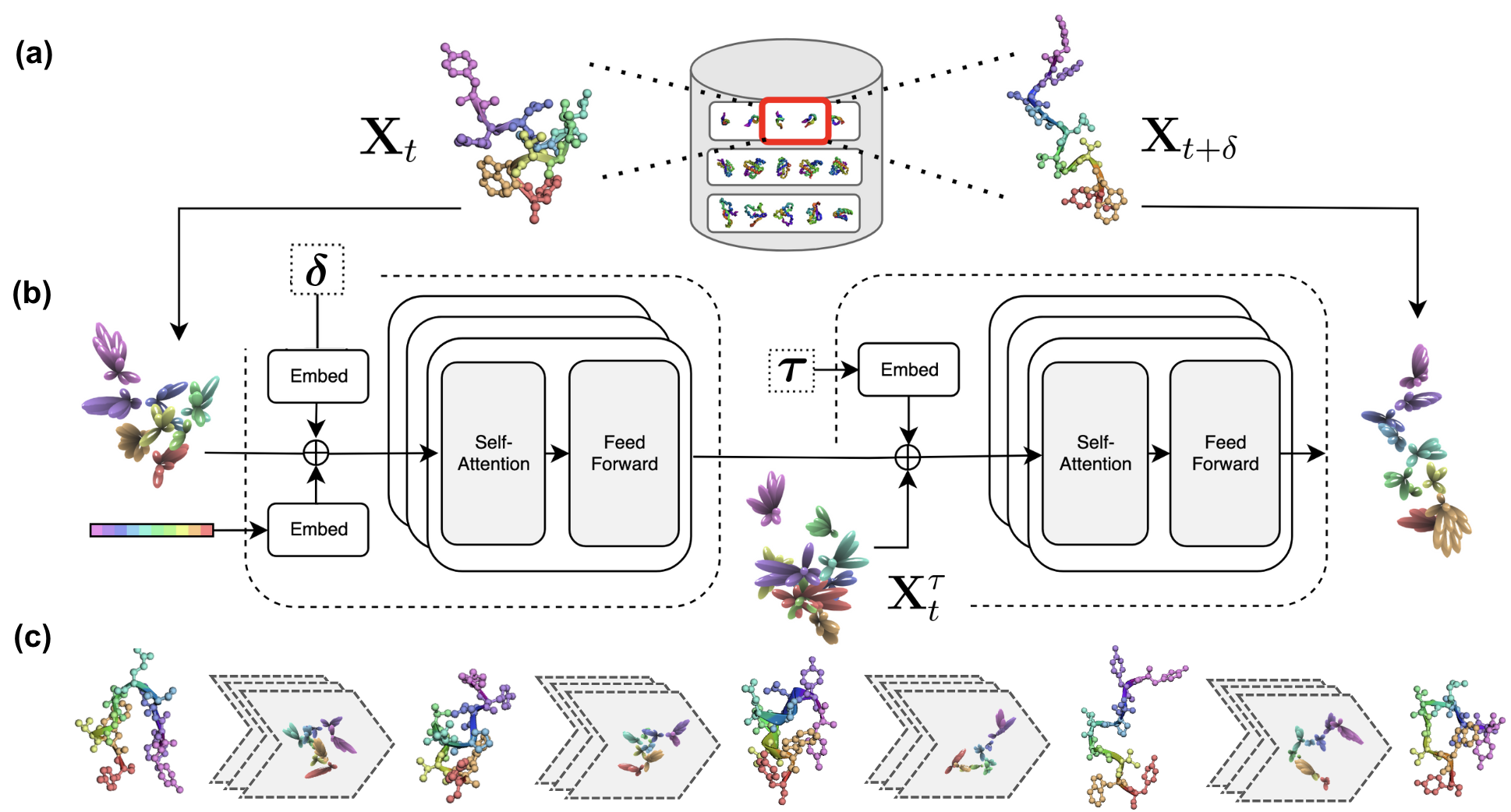
Figure 1: DeepJump architecture and training pipeline, illustrating the use of diverse trajectory data, a two-stage equivariant model, and iterative generative sampling for long protein trajectories.
The generative process is formulated as a conditional flow matching problem, where the model learns to map a noised source state to a future state over a specified time interval. The architecture leverages attention mechanisms adapted for equivariant vector features, and incorporates residual connections and equivariant normalization for stable training. The model is trained with Huber loss on pairwise 3D vectors, and supports multiple model capacities (32, 64, 128 dimensions) to study the effect of scaling.
Generalization to Long-Timescale Protein Dynamics
A central claim of the work is that training on short, structurally diverse trajectories is sufficient to capture generalizable dynamical behavior. To evaluate this, DeepJump is tested on fast-folding proteins with extensive reference MD data, using Time-lagged Independent Component Analysis (TICA) to define macrostates and Markov State Models (MSMs) to quantify dynamical properties. The model is able to reproduce key features of the reference free energy landscapes, including the main conformational basins and transition pathways, with strong agreement in RMSD and fraction of native contacts (FNC) profiles.
Acceleration-Accuracy Trade-offs
A key contribution is the systematic mapping of the trade-off between simulation fidelity and computational speedup as a function of model capacity and jump size δ. Larger jump sizes enable greater acceleration but degrade simulation quality, as measured by stationary distribution distance, folding free energy error, and mean first passage time (MFPT) errors in MSMs. Increasing model capacity partially mitigates this degradation, but does not fully compensate for the loss in accuracy at large jump sizes.
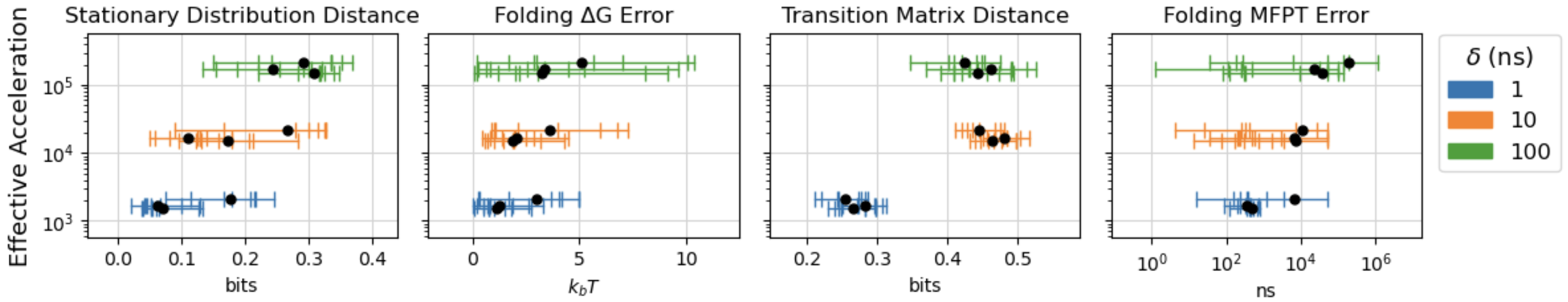
Figure 3: Trade-off between simulation fidelity and computational speedup as a function of model scale and jump size δ.
Quantitatively, DeepJump achieves up to ∼1000× acceleration over classical MD (Amber force-field on A6000 GPU) while maintaining acceptable accuracy for moderate jump sizes and model capacities. For example, with δ=10 ns and 128-dimensional models, stationary distribution distances and folding MFPT errors remain within practical bounds for most fast-folding proteins.
Application to ab initio Protein Folding
DeepJump is further evaluated on ab initio folding tasks, starting from extended β-sheet conformations. The model is able to generate folding trajectories that reach native-like states, with folding success rates and MFPTs comparable to reference MD for small and moderate jump sizes. However, for large jump sizes (δ=100 ns), folding success drops significantly, and the model fails to capture critical intermediate states and rare barrier-crossing events. This highlights the importance of temporal resolution in accurately modeling complex folding pathways.
Model Limitations
Despite its strengths, DeepJump exhibits several limitations. The model fails to generalize to proteins much smaller than those in the training set, producing chemically invalid states. There is a bias toward globular, well-folded conformations, reflecting the composition of the training data. The model is also restricted to standard amino acid residues, limiting its applicability to proteins with non-standard chemistry. These limitations underscore the need for more diverse training data and architectural extensions to handle broader classes of biomolecular systems.
Implications and Future Directions
DeepJump demonstrates that generative, equivariant neural models can learn to accelerate protein MD by orders of magnitude while preserving key dynamical properties. This has significant implications for computational biology, enabling routine simulation of protein dynamics on timescales previously inaccessible to atomistic MD. The explicit mapping of acceleration-accuracy trade-offs provides a framework for selecting model configurations tailored to specific scientific questions, balancing speed and fidelity.
Future developments may include expanding the training dataset to cover a wider range of protein sizes, topologies, and chemistries, incorporating non-standard residues, and integrating adaptive jump size selection. The approach could be extended to other classes of biomolecules, such as nucleic acids or protein-ligand complexes, and combined with experimental data for enhanced realism. Theoretical advances in flow matching and equivariant architectures may further improve the fidelity and generalizability of learned dynamics.
Conclusion
DeepJump establishes a practical, scalable approach for accelerating protein molecular dynamics simulation using generative, equivariant neural networks trained on diverse trajectory data. The model achieves substantial computational acceleration while maintaining fidelity to key dynamical observables, and provides a systematic analysis of the trade-offs inherent in learned MD simulation. These results represent a significant step toward the routine application of machine learning-accelerated simulation in structural biology and related fields.

