- The paper quantifies that inherent genomic entropy undermines transformer attention, producing near-uniform token distributions and unstable embeddings.
- The paper demonstrates through ensemble disagreement and Fisher Information analysis that genomic models lose effective inter-token relationship modeling.
- The paper reveals that directly applying NLP self-supervision to genomics fails unless augmented with domain-specific biological context.
Entropy, Model Disagreement, and Fundamental Constraints of Foundation Models in Genomics
Introduction
The paper "Entropy, Disagreement, and the Limits of Foundation Models in Genomics" (2604.04287) delivers a comprehensive comparative study of foundation models trained on DNA sequences (Genomic Foundation Models, GFMs) versus those trained on natural language. It deploys rigorous architectural and training controls to dissect why GFMs, in contrast to their NLP analogs, demonstrate limited downstream efficacy and interpretability when trained via self-supervised objectives. The central thesis is that the intrinsic entropy of genomic sequences fundamentally constrains model learning, undermines representation stability, and invalidates several assumptions ported from the NLP foundation model paradigm.
Experimental Setup and Methodology
Three ensembles, each of five BERT-style transformer encoders with 90M parameters, underwent training on either tokenized English text or DNA sequences from plant genomes. Both BPE and non-overlapping k-mer tokenizers (k=6) were considered for genomic data, with vocabulary size fixed at 4096, matching the DNA 6-mer space's combinatorial span. Critical to cross-domain comparison, the models were matched for parameter count, training steps (5B tokens), tokenization size, and even the data streaming order, differing only in initial weight randomization.
This stringent setup effectively isolates the statistical properties of the training corpus—especially entropy—as the variable of interest.
Entropy and Output Distribution Flatness
The work quantitatively establishes that genomic sequence entropy, as perceived in the masked prediction setting, far exceeds that of text. This is evidenced by the near-uniformity of token predictive distributions in DNA models: the KL divergence to the uniform distribution for DNA/BPE models is ∼3 bits, dropping below 1 bit for DNA/k-mer models, versus >10 bits for text models. Such distribution flatness indicates profound predictive uncertainty and precludes high-confidence masked token recovery.
Ensemble Disagreement and Representation Instability
To probe model agreement, the Jensen-Shannon (JS) distance between output distributions was computed for model pairs across ensembles. While high-probability mass retention (p close to 1) nominally reduces JS distance, this apparent agreement is illusory; when nucleus sampling lowers p, inter-model distances escalate rapidly among DNA models, revealing deep output instability masked by uniformity.
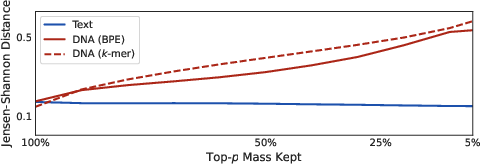
Figure 1: Jensen-Shannon distance E evidences increasing ensemble output disagreement for DNA models under nucleus sampling, while text models remain robustly stable.
Static word embeddings extracted from the first embedding layers further highlight this instability. Metrics such as top-k nearest neighbor Jaccard overlap and Spearman correlation reveal that DNA model embeddings exhibit significantly less inter-model structural agreement, despite training on identical data orders. All DNA models display high variance driven by random initialization, suggesting that the optimization landscape for high-entropy data is highly non-convex w.r.t. meaningful embedding structure. In contrast, text models consistently recover semantically coherent local neighborhoods, aligning with classical static embedding results.
Layer-wise empirical Fisher Information (FIM) provides a parameter sensitivity and identifiability landscape across the network. For text-trained models, Fisher mass aggregates predominantly in transformer layers, reflecting effective capture of inter-token dependencies via the self-attention mechanism.
By stark contrast, DNA-trained models localize Fisher information to the static embedding layer, with little weight in subsequent transformers. This infers that the model learns little about inter-token relationships—the core mechanism behind the transformative power of transformers in NLP is rendered inert by the entropy barrier. The attention mechanism thus fails to find reliable relational signals in DNA, reminiscent of learning by rote association rather than compositionality.
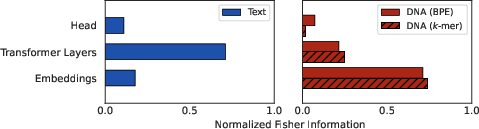
Figure 2: Normalized empirical Fisher information across layer groups demonstrates that text models utilize attention-heavy transformer architecture, whereas DNA models are constrained to embeddings.
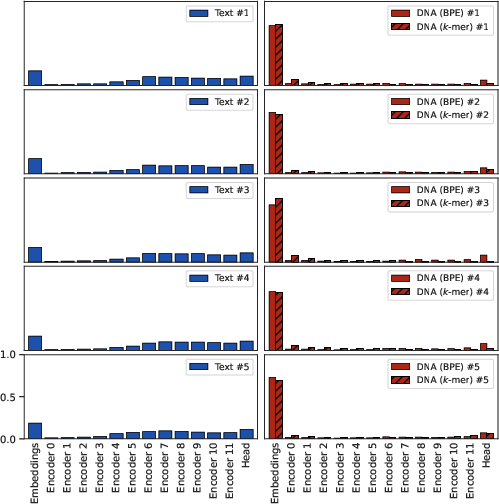
Figure 3: Per-model, per-layer Fisher information breakdown indicates that transformer layers in DNA models are largely information-starved, reinforcing the embedding-centric bottleneck.
Implications for Genomic Foundation Modeling
The study provides robust empirical evidence that the high conditional entropy of genomic data, when seen as token sequences devoid of auxiliary information, generates fundamental obstacles for language-model-derived self-supervision:
- Uncertainty propagation: Output distributions remain flat across contexts, undermining the emergence of distinctive representations required for downstream application generalization.
- Reproducibility risk: Embedding spaces are non-robust with respect to initialization; interpretability and reproducibility within and across training runs are compromised.
- Architectural redundancy: The inductive biases embedded in transformers (attention, context modeling) are not exploited, as Fisher information is funneled into static token identity mappings.
Importantly, attempts to remedy this via alternative tokenizers (e.g., k-mers vs BPE) yield negligible improvement, underscoring that the limitation is data-driven, not model-induced.
Prospects for Future Research
These findings call into question methodologies that transfer the masked-language-modeling paradigm wholesale from NLP to genomics. The theoretical implication is that sequence-level self-supervision, as currently deployed, may not suffice for extracting causal or physically meaningful biological rules from DNA. This result dovetails with recent benchmarking work suggesting that small, specialized models trained directly for downstream tasks consistently outperform large, unsupervised GFMs in genomics.
Mitigating this entropy-induced bottleneck may require:
- Integration of structured biological side information (e.g., epigenomic markers, 3D structure, motif annotations) to reduce effective conditional entropy.
- Architectures incorporating domain-specific inductive biases that transcend sequence-only attention.
- Task-specific supervision or hybrid objectives that force coherent, low-entropy features to emerge in the latent space.
Conclusion
The comparative analysis rigorously demonstrates that the high entropy inherent to DNA sequences is a central limiting factor for developing foundation models analogous to those in NLP. DNA-based self-supervised transformers, even under carefully controlled conditions, exhibit flat prediction distributions, unstable static representations, and ineffective utilization of inter-token context, as validated by both output and parameter-space analyses. These results advocate reevaluating the paradigm for foundation model development in genomics, advocating for approaches that explicitly mitigate entropy-induced uncertainty and leverage domain-specific structure.