PharmAgents: Building a Virtual Pharma with Large Language Model Agents
Abstract: The discovery of novel small molecule drugs remains a critical scientific challenge with far-reaching implications for treating diseases and advancing human health. Traditional drug development--especially for small molecule therapeutics--is a highly complex, resource-intensive, and time-consuming process that requires multidisciplinary collaboration. Recent breakthroughs in AI, particularly the rise of LLMs, present a transformative opportunity to streamline and accelerate this process. In this paper, we introduce PharmAgents, a virtual pharmaceutical ecosystem driven by LLM-based multi-agent collaboration. PharmAgents simulates the full drug discovery workflow--from target discovery to preclinical evaluation--by integrating explainable, LLM-driven agents equipped with specialized machine learning models and computational tools. Through structured knowledge exchange and automated optimization, PharmAgents identifies potential therapeutic targets, discovers promising lead compounds, enhances binding affinity and key molecular properties, and performs in silico analyses of toxicity and synthetic feasibility. Additionally, the system supports interpretability, agent interaction, and self-evolvement, enabling it to refine future drug designs based on prior experience. By showcasing the potential of LLM-powered multi-agent systems in drug discovery, this work establishes a new paradigm for autonomous, explainable, and scalable pharmaceutical research, with future extensions toward comprehensive drug lifecycle management.

Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper introduces PharmAgents, a “virtual pharma” made of smart AI helpers that work together like a real drug company. Their goal is to speed up and explain the early steps of discovering new small‑molecule medicines (tiny chemicals that can become pills). PharmAgents uses LLMs—the kind of AI that understands and writes text—plus specialized science tools to pick good disease targets, design molecules, improve them, and check safety and make‑ability, all on a computer.
What questions were the researchers trying to answer?
- Can a team of LLM “agents” simulate the early drug discovery pipeline from start to finish?
- Can this team make decisions that are sensible, transparent, and useful to human experts?
- Will combining LLMs with scientific tools (for 3D structures, toxicity, and synthesis) produce better candidate drugs than current methods?
- Can the system learn from past attempts and get better over time?
How did they do it?
Think of drug discovery like building a key to fit a biological lock:
- The “lock” is a protein in the body related to a disease.
- The “key” is a small molecule that sticks to (binds) that protein in just the right way.
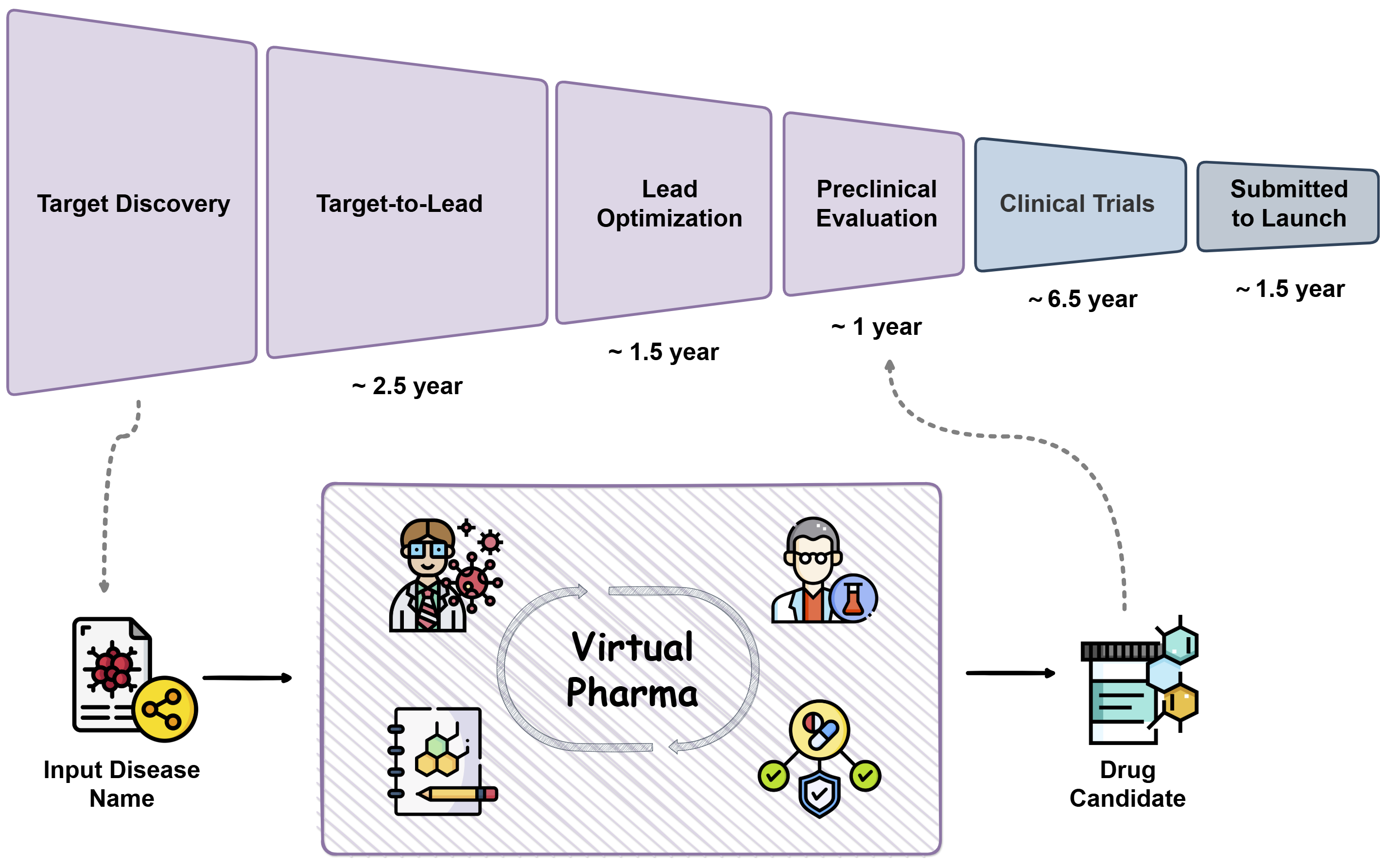
PharmAgents splits this into four stages, each handled by AI agents with different jobs and tools.
1) Target Discovery (finding the right lock)
- A Disease Expert agent looks up diseases and likely protein targets using trusted databases (like UniProt) and its built‑in knowledge from scientific papers.
- A Structure Expert agent finds 3D protein structures (from the Protein Data Bank, PDB) and picks the best versions to work with.
- They use careful filtering steps to avoid bias, keep diversity, and double‑check choices. The result: a short list of protein structures and the exact spot (“pocket”) where a drug should bind.
In simple terms: they choose which protein matters for the disease and where on it the drug should sit.
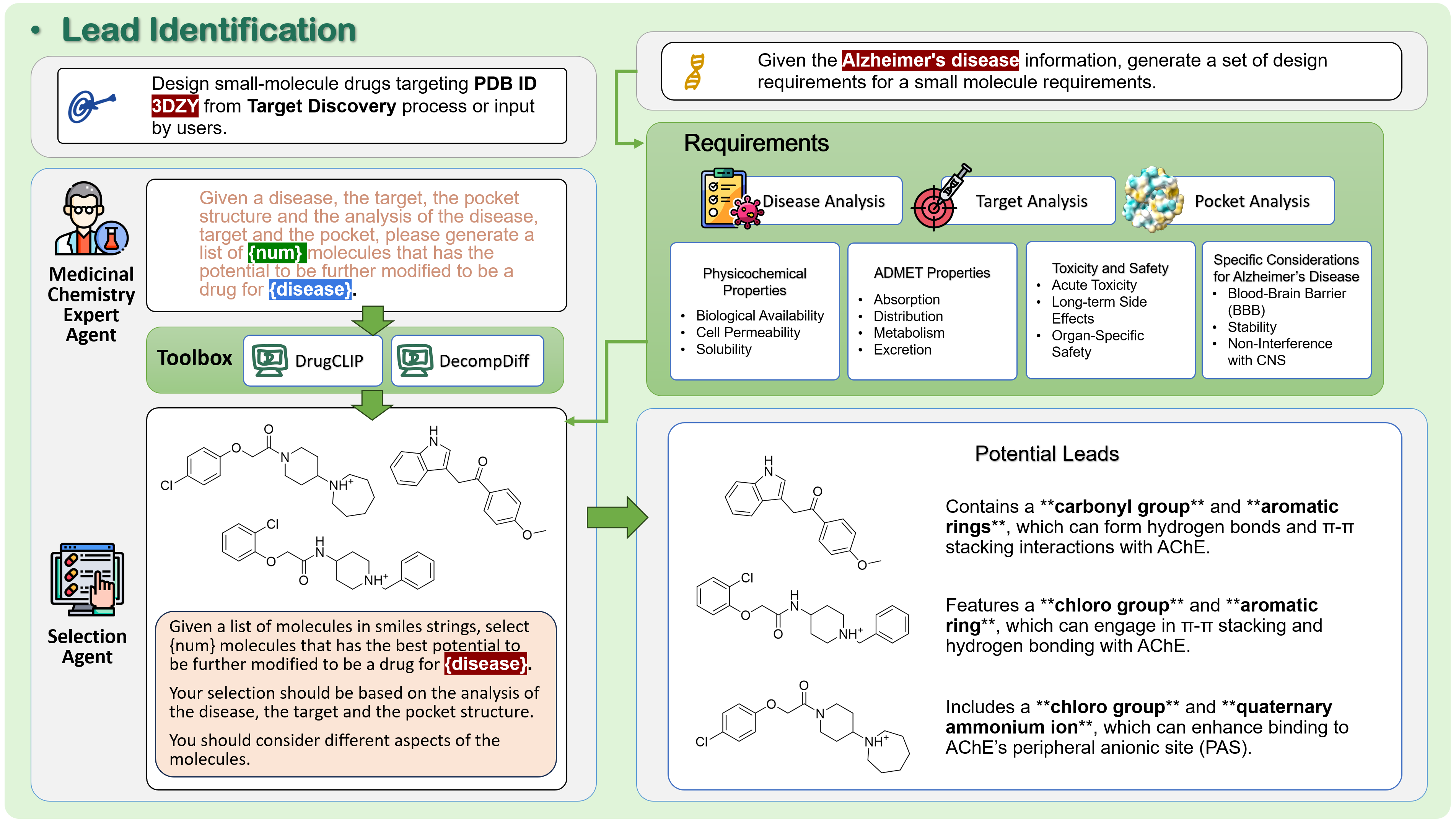
2) Lead Identification (designing a first key)
- Agents analyze the disease and target to write down “wish‑list” properties for molecules. For example:
- For brain diseases: can it cross the blood–brain barrier (enter the brain)?
- For asthma: avoid entering the brain to reduce side effects.
- Three ways to find starting molecules:
- Structure‑based generator (like DecompDiff) that designs molecules to fit the pocket shape.
- Virtual screening (like DrugCLIP) that searches huge libraries for likely binders.
- LLM‑guided design, where the AI proposes molecules using what it learned from the analyses.
- A selection agent picks the best set that matches the wish‑list.
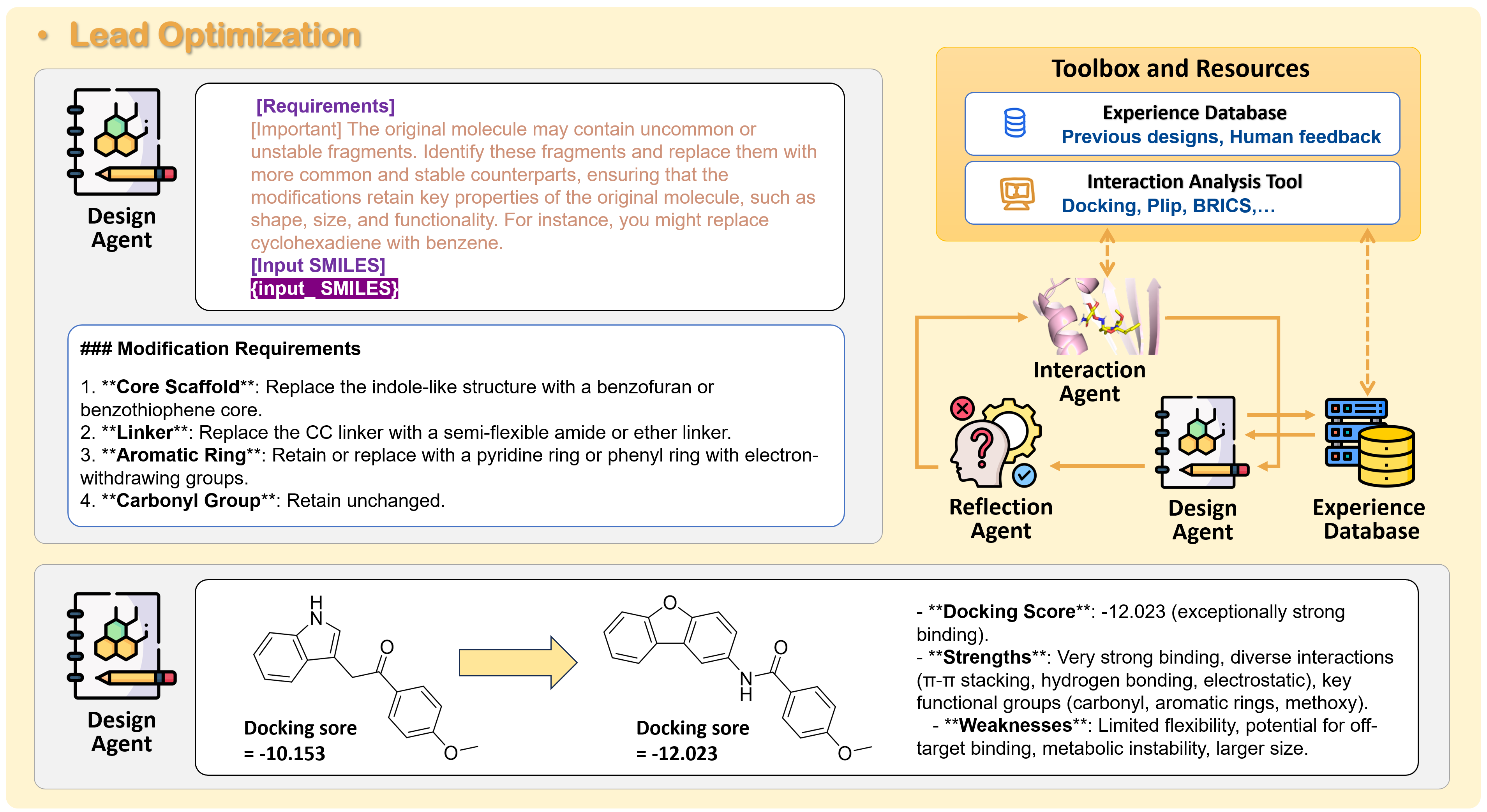
3) Lead Optimization (improving the key)
- Docking tools “try the key in the lock” in 3D and score how well it fits (binding affinity).
- A tool called PLIP reads the protein–molecule contact map—like noting all the “handshakes” (hydrogen bonds, hydrophobic contacts).
- A Design Agent suggests small changes to the molecule to improve fit and drug‑likeness (being realistic to make, stable, and safe).
- A Generation Agent applies those changes to create new versions.
- A Reflection Agent reviews progress and guides the next round.
- This loop runs several times to polish the molecule.
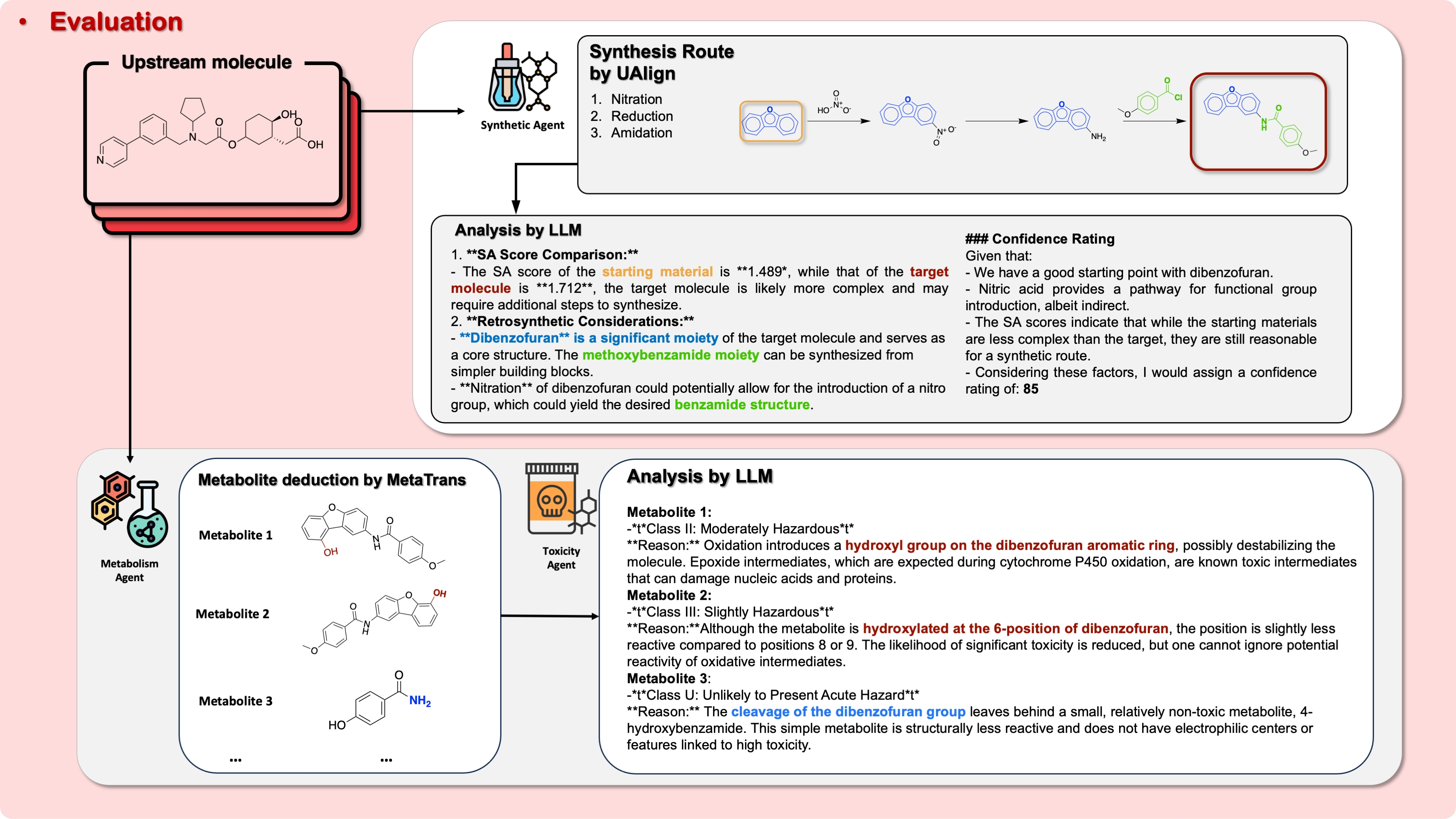
4) Preclinical Candidate (PCC) Evaluation (safety and make‑ability checks)
- Metabolism & Toxicity:
- A model (MetaTrans) predicts how the body might break the molecule into metabolites.
- Toxicity is estimated by comparing “chemical fingerprints” (think barcodes for molecules) to known toxic/less‑toxic chemicals and reasoning with an LLM. Lower risk is better.
- Synthesis (can we make it?):
- Retrosynthesis (with UAlign) plans a recipe backward from the molecule to simple starting materials—like rewinding a cooking video to list ingredients.
- The system scores how practical it is to make the molecule.
- A final agent reads all reports and decides if a molecule is promising enough to move forward.
What’s special about using LLM agents?
- Explainability: They don’t just give scores; they explain their reasoning in readable language.
- Teamwork: Each agent has a role, and they talk to each other to avoid mistakes piling up.
- Self‑learning: The system keeps an experience database—lessons from past designs help future ones.
What did they find?
- Target picking worked well:
- In one test disease, 16 out of 18 chosen protein structures were judged appropriate by human experts.
- The system correctly highlighted important targets for atopic dermatitis (like JAK1), matching real‑world treatments.
- Better molecules:
- Across tests, the success rate (meeting all key property goals at once) rose from 15.72% to 37.94% compared to strong baseline methods—about a 3× improvement.
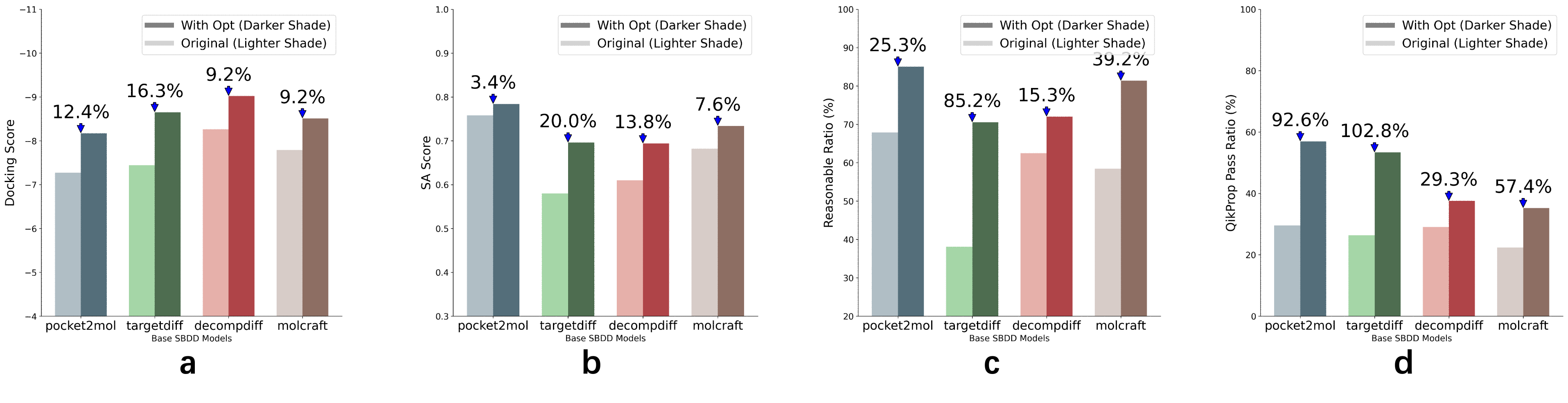
- Optimization improved multiple metrics, such as:
- Binding scores (how tightly the molecule fits the protein)
- Reasonable chemistry (not weird or unstable)
- Synthesizability (how doable the recipe is)
- Drug‑likeness (property checks used in pharma)
- Safer, more practical picks:
- Toxicity underestimation risk was kept low (12%).
- Synthesis evaluations aligned well with quantitative scores (Pearson correlation 0.645), and the system could explain why.
- It learns:
- Using past experience improved success from 30% to 36% in new projects.
Why is this important?
- Faster and cheaper starts: Early drug discovery is slow and expensive. A virtual, explainable system can explore more ideas quickly and narrow down better candidates for lab testing.
- Human–AI teamwork: Clear reasoning builds trust, so scientists can understand and verify AI suggestions.
- Smarter decisions: Combining language understanding with 3D chemistry, safety, and synthesis tools helps balance many needs at once—fit, safety, and buildability.
- A path to scalable R&D: With multi‑agent collaboration and self‑learning, this approach could grow into broader drug lifecycle support.
What are the big takeaways?
- PharmAgents shows that a coordinated team of LLM agents plus science tools can simulate a large part of early drug discovery in a clear, step‑by‑step way.
- It not only finds promising molecules but also explains why they might work, how to improve them, and whether they’re safe and practical to make.
- While real lab and clinical trials are still essential, this virtual pharma could save time, reduce costs, and help researchers focus on the most promising options.
Collections
Sign up for free to add this paper to one or more collections.