MADD: Multi-Agent Drug Discovery Orchestra
Abstract: Hit identification is a central challenge in early drug discovery, traditionally requiring substantial experimental resources. Recent advances in artificial intelligence, particularly LLMs, have enabled virtual screening methods that reduce costs and improve efficiency. However, the growing complexity of these tools has limited their accessibility to wet-lab researchers. Multi-agent systems offer a promising solution by combining the interpretability of LLMs with the precision of specialized models and tools. In this work, we present MADD, a multi-agent system that builds and executes customized hit identification pipelines from natural language queries. MADD employs four coordinated agents to handle key subtasks in de novo compound generation and screening. We evaluate MADD across seven drug discovery cases and demonstrate its superior performance compared to existing LLM-based solutions. Using MADD, we pioneer the application of AI-first drug design to five biological targets and release the identified hit molecules. Finally, we introduce a new benchmark of query-molecule pairs and docking scores for over three million compounds to contribute to the agentic future of drug design.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper introduces MADD, which stands for Multi-Agent Drug Discovery Orchestra. Think of it like a smart team of AI “helpers” that work together to design and check new drug-like molecules based on simple text instructions from a scientist. The goal is to speed up the early stages of drug discovery—finding “hit” molecules that look promising—while making advanced AI tools easier to use for lab researchers.
What questions does the paper try to answer?
The researchers ask a simple but important question: If we split the complex job of finding new drug molecules among several specialized AI agents, can we do a better job than using just one AI model?
More specifically, they investigate:
- Can a multi-agent system understand a scientist’s text request, generate suitable molecules, and evaluate those molecules’ properties end-to-end?
- Will such a system be more accurate and efficient than existing AI tools?
- Can it work even on new disease targets where it hasn’t been set up ahead of time?
How did they do it?
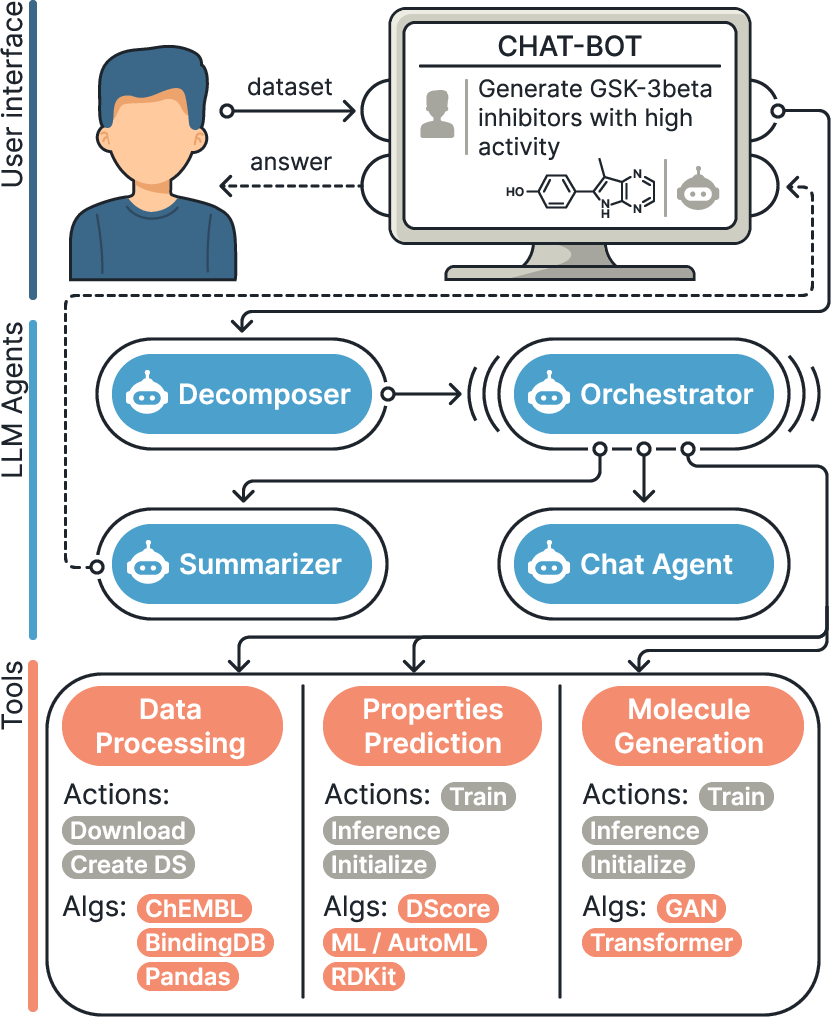
They built MADD as a “team” of four AI agents plus specialized tools. Here’s how it works, using everyday comparisons:
- Decomposer agent: Like a teacher breaking a big homework problem into smaller steps, it takes the user’s request and splits it into manageable tasks.
- Chat agent: Like a helpful tutor, it asks for clarification if the request is unclear and helps the user phrase their needs.
- Orchestrator agent: Like a project manager, it decides which specialized tools to use, in what order, to generate molecules and check their properties.
- Summarizer agent: Like a report writer, it gathers all the results and presents them clearly.
Behind the scenes, MADD uses two kinds of tools:
- Generators (to create molecules): These include models like GANs and Transformers. Think of them as “molecule composers” that write new chemical structures using a kind of chemical alphabet called SMILES.
- Property predictors (to score molecules): These estimate how good a molecule might be, using measures like:
- Docking score: An estimate of how tightly a molecule might fit into a target protein, like how well a key fits a lock. Lower (more negative) is better here.
- IC50: How much of the drug it takes to block half of a target’s activity—like how much volume you need to turn down to halve the noise. Lower is usually better.
- SA (synthetic accessibility): How easy it might be to make the molecule in a lab.
- QED (drug-likeness): Overall “drug-friendliness,” based on several traits.
- Structural filters (PAINS, Brenk, Glaxo, SureChEMBL): Simple checks to remove molecules likely to be troublesome or misleading.
They also use AutoML, which is like an automatic coach that trains and picks the best machine learning models for a given task without needing a human to tune every detail. And they bring in data from trusted sources like ChEMBL and BindingDB to train and test the system.
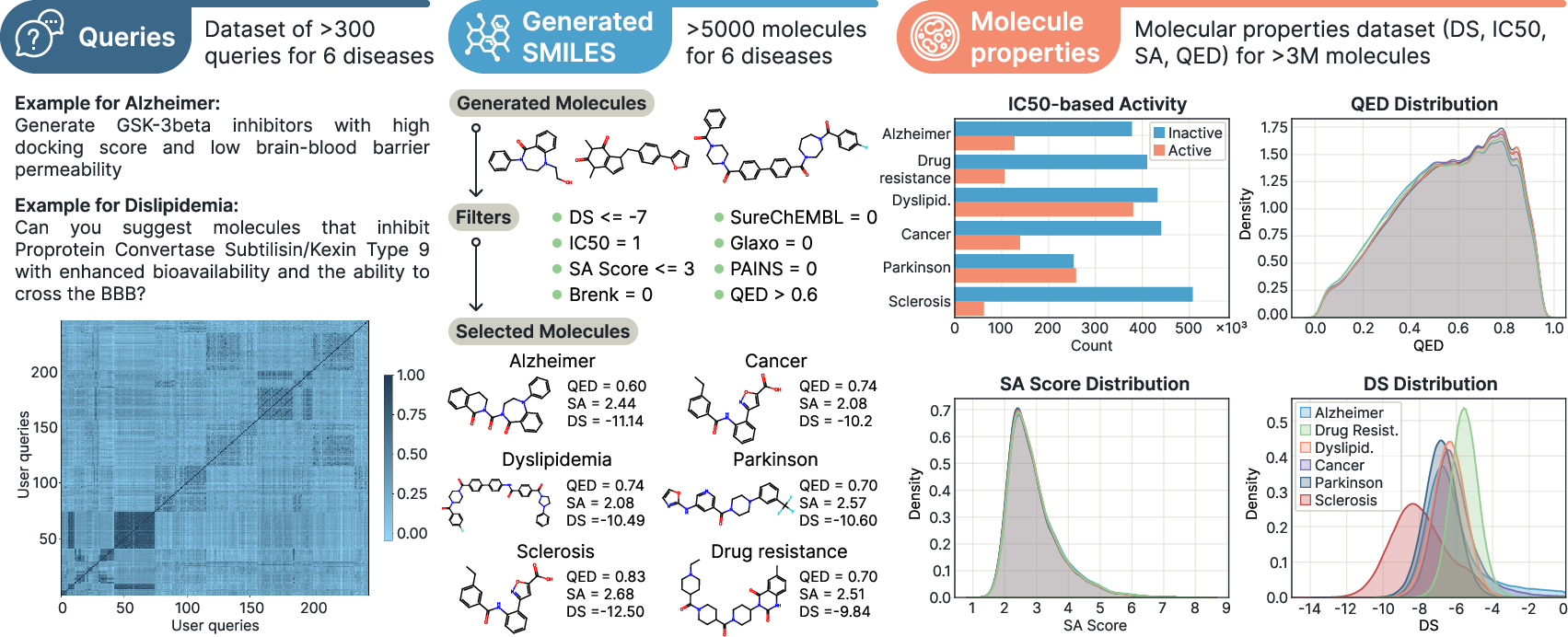
Finally, they created a new benchmark: lots of realistic text queries and millions of molecules with scores, so future teams can compare their systems fairly.
What did they find, and why does it matter?
Here are the main results, explained simply:
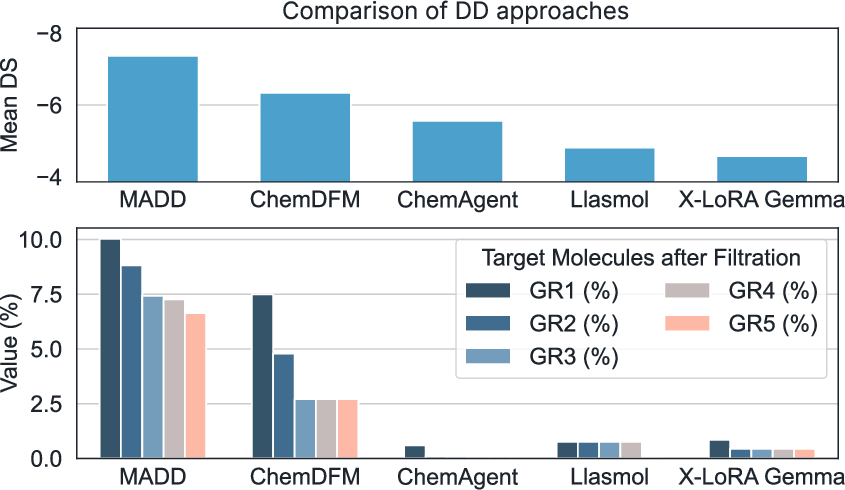
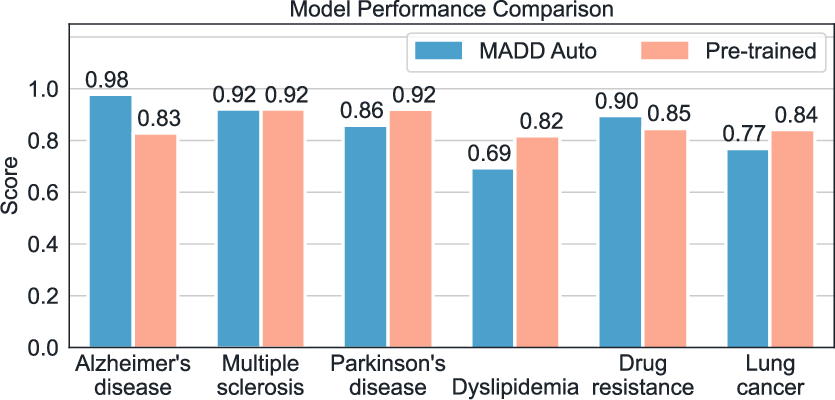
- Multi-agent beats single-agent: Splitting the work across specialized agents made the system more reliable and clearer in its final answers. MADD achieved about 79.8% “final accuracy” on complex, multi-step tasks—much higher than several competing systems.
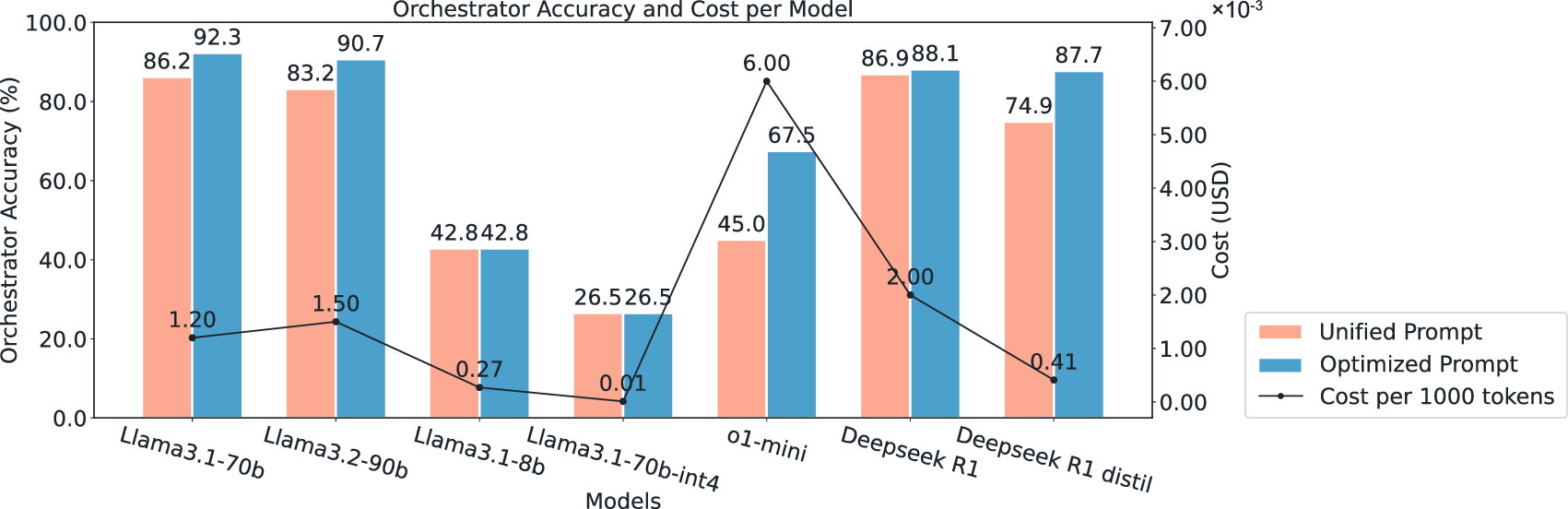
- Better orchestration: They tested lots of LLMs for the “Orchestrator” role and found that a specific model (Llama‑3.1‑70b, with tuned prompts) picked the right tools most accurately.
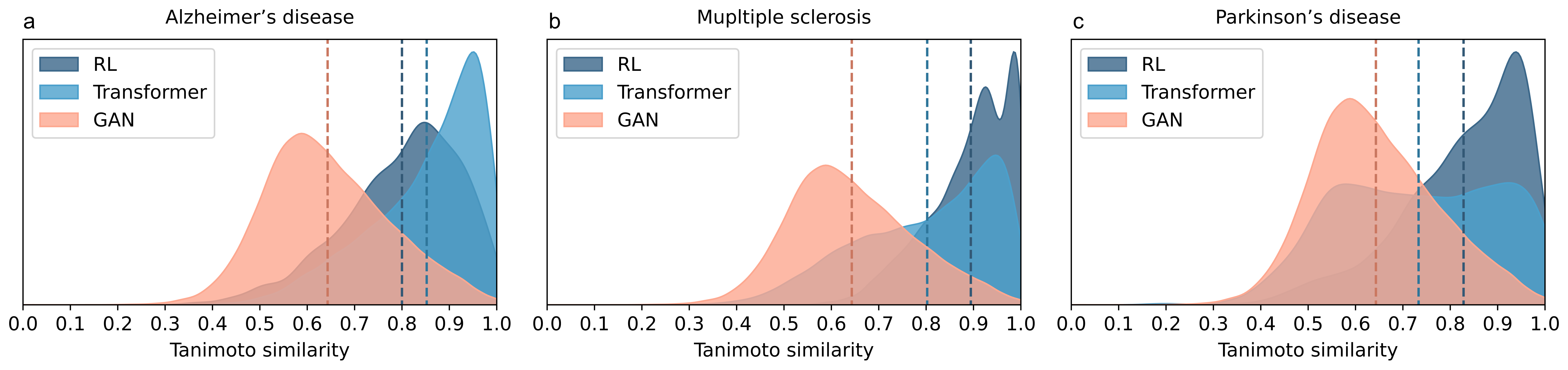
- Strong molecule generation: MADD’s Transformer-based generator produced more “hit” molecules (ones meeting strict filters) across several disease cases compared to other methods. GAN was fast and solid, while Transformer was consistently strong.
- Competitive vs. popular tools: Systems that rely on a single LLM struggled to produce valid, useful molecules. Another agent system (ChemAgent) made many output mistakes—MADD avoided these and delivered clearer, more complete results.
- AutoML helps: The automated training in MADD often outperformed manually trained models for property prediction (like IC50). It also trained generators effectively, making the pipeline more self-sufficient.
- Real-world case studies:
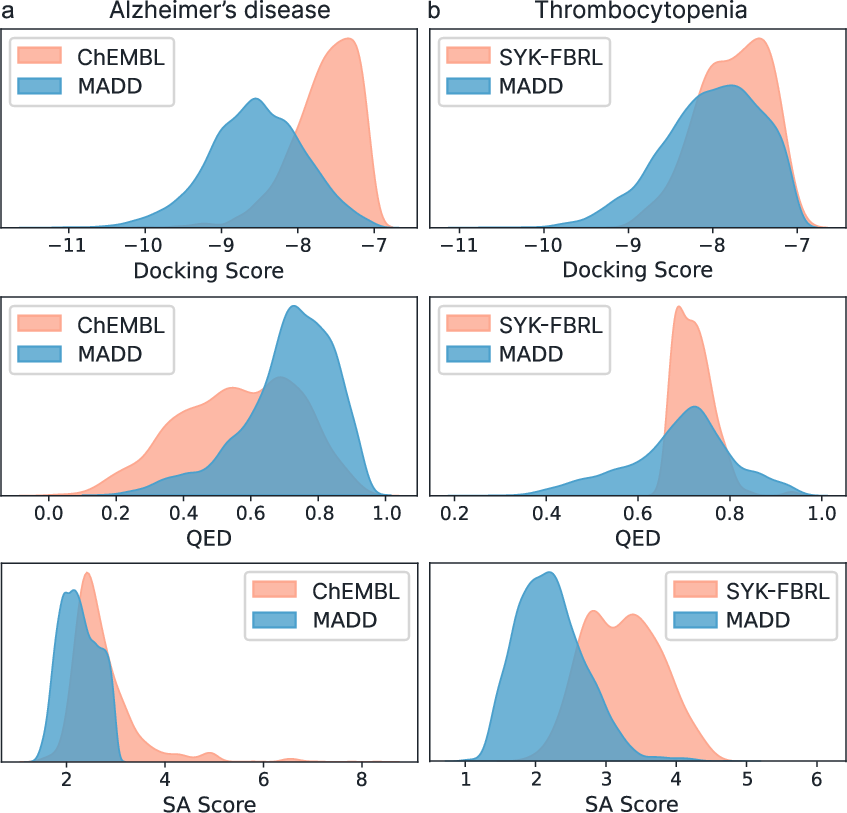
- Alzheimer’s disease: MADD generated molecules that, on average, looked easier to make and more drug-like (higher SA and QED) and showed better binding scores than known reference inhibitors.
- Thrombocytopenia (new/unseen case): MADD automatically trained models using ~3,200 known molecules, then generated 10,000 new molecules. Even with fewer total molecules than a previous method, MADD found a comparable number of top hits and, on average, better scores—showing strong efficiency.
Why this matters: Early drug discovery is costly and slow. MADD shows that an “AI-first” multi-agent system can understand natural language requests, propose new molecules, and judge their promise—all with minimal setup—potentially saving time, money, and effort.
What are the bigger implications?
- Easier access to advanced AI: Wet-lab researchers don’t need to be AI experts—MADD can translate natural-language goals into end-to-end pipelines, which could democratize early drug design.
- Faster, cheaper screening: Generating and scoring molecules virtually can reduce the number of compounds that need expensive lab tests.
- Strong foundation for future tools: The released code and datasets (including millions of docking scores and curated query sets) help the wider community build better systems.
At the same time:
- You still need lab work: Computer predictions are just the first step. Promising molecules must be tested in real biochemical experiments to confirm they really work.
- Data and targets matter: For new diseases, you often need good training data and some idea of the biological target. MADD works best when that information is available.
- Transparency and integration: Future versions could make the system’s decisions more visible and make it simpler to add new tools.
Bottom line
MADD shows that a carefully designed multi-agent AI “orchestra” can turn plain-English drug design ideas into tested molecular suggestions, often outperforming existing approaches. It won’t replace the lab, but it can make the journey from idea to testable candidates smoother, quicker, and more accessible.
Knowledge Gaps
Below is a consolidated list of concrete knowledge gaps, limitations, and open questions the paper leaves unresolved. Each point is phrased to enable actionable follow-up by future researchers.
- Lack of prospective wet‑lab validation: no biochemical, cell-based, or in vivo assays are reported to confirm predicted binding, efficacy, or safety; define a plan for iterative design–test–learn cycles and selection criteria for advancing molecules to assays.
- Heavy reliance on docking scores and predicted IC50 without cross-validation: no comparison across multiple docking engines, receptor ensembles, or post‑docking rescoring (e.g., MM‑GBSA, FEP); quantify how hit rates change with protein flexibility, protonation/tautomer states, and binding-site water/cofactor treatment.
- Missing uncertainty quantification and calibration: predictions (IC50, docking) lack confidence intervals, calibration curves, or error bars; incorporate UQ to prioritize candidates with high confidence and identify unreliable regions of chemical space.
- No ADMET/toxicity integration: beyond QED and structural filters, the pipeline omits predictive models for hERG, CYP interactions, solubility, permeability, metabolic stability, and hepatotoxicity; assess how adding ADMET predictors affects hit quality and attrition rates.
- Synthetic feasibility not grounded in reaction planning: SA scores are used, but no retrosynthesis planning, building-block availability, reaction-condition constraints, or cost/yield estimation are considered; integrate synthesis planning tools (e.g., ASKCOS/AiZynthFinder) and validate route feasibility.
- Off‑target risk and selectivity unaddressed: no models for selectivity across homologs or known liability targets; add multi-target screening to reduce off-target effects and improve clinical relevance.
- Multi‑objective optimization strategy is simplistic: filtering thresholds act as hard gates, but trade‑offs (e.g., potency vs. ADMET vs. synthesizeability) are not optimized; implement Pareto frontiers and explicit weighted objectives to balance competing properties.
- Generalization to truly novel targets with little/no data: current workflow requires user-supplied datasets and prior target knowledge; explore zero-shot/transfer methods, protein LLMs, pocket-conditioned generation, and automated hypothesis generation for new targets.
- Benchmark representativeness and ground truth: the query dataset is semi-synthetic (seeded by ~30 manual queries and augmented via LLMs) and lacks ground-truth mappings from queries to known hits; conduct user studies with wet‑lab scientists and add tasks with established reference solutions.
- Evaluation metrics may not reflect real hit utility: TS/SSA/FA measure orchestration and summarization, but their correlation with chemical hit rates and downstream lab success is unknown; perform correlation and ablation analyses linking agent metrics to molecular outcomes.
- Comparative baselines may be mismatched: standalone LLMs lack tool access while ChemAgent uses different orchestration; include stronger structure‑based and 3D generative baselines (e.g., diffusion models, pocket-conditioned generators) and harmonize tool access, prompts, and evaluation criteria.
- Filter threshold selection is opaque and user‑dependent: thresholds for docking, IC50, and filters (Brenk, PAINS, SureChEMBL, Glaxo, QED) are target/context‑dependent; provide automatic, target‑specific calibration and sensitivity analysis to justify chosen cutoffs.
- Protein structure choices and preparation are unspecified: the paper does not detail PDB selection, binding site definition, co-factors, mutations, protonation, or ensemble usage; standardize protein preparation and report protocol variability impacts on docking outcomes.
- Molecule 3D realism and enumeration are not considered: generation is SMILES‑based without stereochemistry enforcement, tautomer/protomer enumeration, conformer generation, or 3D property checks; quantify effects of proper 3D handling on docking and property predictions.
- Data quality, leakage, and novelty criteria need auditing: ensure strict train/test separation for generative/predictive models, extend novelty checks beyond internal datasets to broader prior art, and report duplication/removal protocols.
- Scalability and cost are not characterized: no breakdown of runtime, GPU/CPU requirements, energy cost, or per‑query expenditure for agents, docking, training, and screening; provide performance–cost profiles and scaling limits under concurrent use.
- Tool integration and extensibility barriers: adding new tools requires code changes; introduce a standardized plugin API, declarative tool schemas, and configuration-driven integration to enable non‑programmers and accelerate ecosystem growth.
- Agent robustness to language variation is unknown: the system is only evaluated on English queries; test multilingual inputs, domain‑specific jargon, noisy/ambiguous instructions, and non‑Latin scripts, and quantify TS/SSA/FA degradation.
- Memory, planning, and reproducibility: no analysis of long‑horizon task memory or reproducible execution; add agent decision logs, deterministic seeds, and provenance tracking for full auditability of pipelines and outputs.
- Continual learning and model updates: how to update predictive/generative models over time without catastrophic forgetting or drift is not defined; design MLOps for periodic retraining, validation gates, and rollbacks.
- Human‑in‑the‑loop strategies are missing: there is no protocol for incorporating medicinal chemist feedback, active learning from assay results, or preference/rationale guidance to refine generations; evaluate how expert input changes hit rates and diversity.
- Safety and misuse risks need stronger guardrails: beyond a mention of “risks,” there is no red‑teaming or automated filters for toxic/opioid/chemical weapons precursors; formalize policy checks and dual‑use detection in generation and summarization steps.
- Patentability and IP diligence are not assessed: novelty is reported relative to limited datasets, but patent prior‑art searches and freedom‑to‑operate analyses are absent; integrate IP screening to prioritize patentable and non‑encumbered scaffolds.
- Modalities beyond small molecules: peptides, macrocycles, covalent inhibitors, PROTACs, and RNA ligands are not supported; quantify how extending to these modalities changes orchestration, tools, and validation pipelines.
- Real‑world usability and adoption: no user studies with wet‑lab researchers to assess clarity of outputs, trust, and decision utility; measure perceived usefulness, error modes, and training needs in practical settings.
- Failure mode taxonomy and mitigation: while an appendix mentions failures, there is no quantitative classification or mitigation strategies; build a failure registry, root‑cause analysis, and automated recovery policies for agent/tool missteps.
- Impact of automatically adding training tools: TS drops when integrating training; investigate planning policies (e.g., tool selection constraints, curriculum scheduling) to maintain accuracy while enabling AutoML.
- Orchestrator choice sensitivity: Llama‑3.1‑70b is best in this study, but sensitivity to model updates, distillation to smaller models, and cost-performance trade‑offs remain open; evaluate portability to lighter LLMs and on‑prem deployments.
- External reproducibility of docking scores in the released 3M benchmark: clarify docking engine versions, parameters, protein preparation protocols, and reproducibility across platforms to ensure reliable benchmarking by third parties.
Practical Applications
Practical Applications Derived from the MADD Paper
This paper presents MADD, a multi-agent “orchestra” for early-stage drug discovery that transforms natural-language queries into end-to-end, target-adaptive hit identification pipelines. It integrates LLM-based orchestration, generative models (GAN, Transformer/CVAE), predictive models (IC50, docking), AutoML for model training, and cheminformatics filters (SA, QED, Brenk/SureChEMBL/Glaxo/PAINS). The open-source code and a new benchmark (queries + docking for >3M molecules) enable both immediate deployment in R&D workflows and longer-term development of autonomous, closed-loop discovery systems.
Immediate Applications
Below are actionable use cases that can be deployed now with the paper’s released code, datasets, and demonstrated workflows.
- Sector: Biopharma R&D (industry, CROs)
- Application: AI-first hit identification assistant for known targets
- What it delivers: Query-to-hit pipelines that generate and triage candidate molecules against a specified protein (e.g., STAT3, ABL, COMT, ACL, PCSK9), with computed docking scores, IC50 predictions, SA, QED, and structural alert filtering.
- Tools/products/workflows: MADD’s four agents (Decomposer, Orchestrator, Summarizer, Chat Agent), AutoML-DL, DatasetBuilder, RDKit filters; standard “GR1–GR5” multi-criteria filters; top-N hit lists with structured summaries for chemist review.
- Assumptions/dependencies: Known target and access to activity/docking data (via BindingDB/ChEMBL or internal assays), GPU/CPU compute for docking/modeling, cheminformatics stack (RDKit), and tolerance for in silico proxies pending wet-lab validation.
- Sector: CRO services
- Application: “Agentic virtual screening” offering
- What it delivers: A fee-for-service pipeline where clients submit a target and constraints; the CRO uses MADD to build datasets, train predictors, fine-tune generators, and return validated hit lists and purchase/synthesis suggestions.
- Tools/products/workflows: DatasetBuilder-driven data ingestion, AutoML training with FEDOT, MADD orchestration and summarization, deliverable packs (SMILES, docking poses/scores, SA/QED/alerts, novelty metrics).
- Assumptions/dependencies: Data-use agreements; secure multi-tenant deployment; integration with synthesis vendors or in-house chemistry for follow-up.
- Sector: Academic labs and teaching
- Application: Rapid hypothesis testing and method education
- What it delivers: A lightweight path for non-specialist wet-lab groups to turn textual hypotheses into candidate lists; classroom exercises/assignments using the released benchmark to teach agentic AI for chemistry.
- Tools/products/workflows: Prebuilt prompts and workflows; open benchmark (query sets + docking over >3M molecules) and code to reproduce the paper’s cases (e.g., Alzheimer’s, Parkinson’s, MS, lung cancer, dyslipidemia, drug resistance).
- Assumptions/dependencies: Minimal compute for small pilot runs; institutional access to docking if scaling; basic Python environment.
- Sector: Software for R&D (ELN/LIMS/SDMS vendors)
- Application: “Chat-to-pipeline” plugin inside lab informatics
- What it delivers: A UI widget that lets scientists type target/property intents and get back structured hit lists, with audit of tools used.
- Tools/products/workflows: MADD as a backend service; REST/gRPC adapter; result cards with traceable tool calls and filters applied.
- Assumptions/dependencies: IT/security approvals; containerized deployment; controlled model hosting (Llama-3.1-70B or alternative).
- Sector: Portfolio triage (biotech, venture studios)
- Application: Computational triage of large libraries for a program/asset
- What it delivers: Fast prioritization of internal or commercial libraries against program targets, producing a shortlist for purchase/synthesis.
- Tools/products/workflows: Batch docking and ML property prediction, GR1–GR5 gating, novelty and diversity metrics (e.g., Tanimoto).
- Assumptions/dependencies: HPC budget for large screens; licensing for docking engines; compound availability mapping.
- Sector: Methods research (academia/industry ML groups)
- Application: Benchmark-driven research on agent orchestration
- What it delivers: A public benchmark for multi-agent drug design (query-to-hit) to evaluate tool selection accuracy, summarization, and end-to-end outcomes; ablation baselines (e.g., demonstrating the value of a dedicated Summarizer).
- Tools/products/workflows: The released benchmark, MADD’s modular agents, orchestration accuracy (OA/TS), SSA/FA metrics.
- Assumptions/dependencies: Reproducible compute; adherence to benchmark protocols.
- Sector: Education/outreach
- Application: Safe demo of property-aware molecule generation
- What it delivers: A web demo for learners to submit benign chemistry queries and observe molecule generation with built-in structural alerts and drug-likeness checks.
- Tools/products/workflows: MADD with stricter filters and rate limits, curated example prompts, safety guardrails (PAINS/Glaxo/Brenk).
- Assumptions/dependencies: Governance to prevent misuse; restricted model capabilities for public access.
- Sector: Internal compliance and documentation (regulated R&D)
- Application: Experiment traceability for computational hit finding
- What it delivers: Traceable logs of agent decisions, tool calls, datasets used, and parameters to support internal reviews and knowledge capture.
- Tools/products/workflows: MADD’s modular prompts and pipelines plus added decision logging; standardized result reports for SOP archives.
- Assumptions/dependencies: Additional engineering for full audit trails; alignment with GxP-aligned documentation standards.
- Sector: Target-focused library design for ordering/synthesis
- Application: Vendor-orderable hit lists
- What it delivers: Shortlists of high-SA and high-QED candidates mapped to commercial catalogs or close analogs, ready to order or synthesize.
- Tools/products/workflows: MADD generation + SA/QED filtering; similarity search against vendor catalogs; procurement-ready export.
- Assumptions/dependencies: Access to supplier databases; tolerance for analog-based selection where exact hits are not purchasable.
- Sector: Community and open science
- Application: Leaderboards and shared tasks for agentic drug design
- What it delivers: Public challenges using the provided queries and docking data to accelerate reproducibility and best practices.
- Tools/products/workflows: Leaderboard backend; evaluation scripts for TS/SSA/FA and GR1–GR5 hit rates.
- Assumptions/dependencies: Community stewardship; compute sponsorship for participants.
Long-Term Applications
These use cases require further research, scaling, integration with wet-lab automation, or regulatory maturation.
- Sector: Pharma R&D and robotics
- Application: Closed-loop design–make–test–learn (DMTL)
- What it could deliver: Autonomous campaigns where MADD proposes molecules, passes them to synthesis planning/robotic platforms, triggers assays, and retrains models—shrinking cycle times.
- Tools/products/workflows: Integration with synthesis planners (e.g., retrosynthesis/ASKCOS), automated chemistry (chemputers), bioassay robotics, ELN/LIMS; continual learning loops.
- Assumptions/dependencies: Robotic lab access, standardized APIs, robust experiment data capture, budget for capital equipment.
- Sector: Drug discovery for novel/poorly characterized targets
- Application: Hypothesis generation when targets and assays are nascent
- What it could deliver: Modules for literature/RAG knowledge extraction, homology modeling, and multi-modal evidence synthesis to propose initial target hypotheses and ligandable sites, feeding MADD’s generation pipeline.
- Tools/products/workflows: Knowledge graphs, structure prediction (e.g., AlphaFold-like), target deconvolution; agent chains for evidence grading.
- Assumptions/dependencies: Quality/coverage of public literature and structures; validation with bespoke assays.
- Sector: Clinical translation enablement
- Application: Multi-objective design with ADMET/PK/PD and off-target profiles
- What it could deliver: Agents that co-optimize efficacy, safety, developability, and liability risks; automated trade-off exploration.
- Tools/products/workflows: ADMET predictive ensembles, PBPK/PK/PD models, polypharmacology/off-target screens; Pareto-front exploration.
- Assumptions/dependencies: Robust, unbiased ADMET datasets; careful calibration and prospective validation.
- Sector: Rapid-response discovery (public health, emerging threats)
- Application: Time-bound hit finding for outbreaks/novel pathogens
- What it could deliver: 24–72-hour campaigns to propose testable hits against emergent targets (e.g., viral proteins), prioritizing makeability and novelty.
- Tools/products/workflows: Fast docking pipelines, few-shot AutoML, templated prompts/workflows for outbreak modes; synthesis partnerships on standby.
- Assumptions/dependencies: Rapid access to target structures or homology models; standing compute; pre-negotiated synthesis/test capacity.
- Sector: Regulatory science and policy
- Application: Good AI Practice (GxAI) frameworks and AI-native submissions
- What it could deliver: Accepted standards for documenting agentic pipelines (decision logs, datasets, models), validation reports, and traceability to support regulatory dossiers.
- Tools/products/workflows: Agent decision logging, model cards, data lineage, reproducibility kits; third-party audits/benchmarks (e.g., using the MADD benchmark).
- Assumptions/dependencies: Regulator–industry–academia coordination; standard-setting bodies; legal clarity on IP for AI-generated molecules.
- Sector: Personalized/stratified therapeutics
- Application: Variant-aware or subtype-specific small-molecule design
- What it could deliver: Compounds optimized for molecular subtypes (e.g., mutational status) or resistance profiles, guided by patient-derived omics.
- Tools/products/workflows: Integration of genomic/transcriptomic biomarkers into property objectives; virtual patient cohorts for in silico stratification.
- Assumptions/dependencies: Access to high-quality patient data; privacy safeguards; clinical validation pathways.
- Sector: Multi-domain chemical design (materials, agrochemicals)
- Application: Porting the agentic architecture to other chemical sectors
- What it could deliver: Natural-language-to-design pipelines for catalysts, polymers, or crop-protection agents with multi-objective property targets.
- Tools/products/workflows: Sector-specific generators/predictors, alternate property calculators, new filters and datasets.
- Assumptions/dependencies: Domain datasets of sufficient size/quality; sector-relevant physics/ML toolchains.
- Sector: Finance and strategic planning
- Application: Data-driven program valuation and due diligence
- What it could deliver: Early risk/return assessment using predicted hit rates, novelty/diversity, and makeability to inform investment and portfolio decisions.
- Tools/products/workflows: Analytics dashboards fed by MADD outputs; comparators to historical hit-to-lead conversions.
- Assumptions/dependencies: Correlation of in silico metrics with historical outcomes; access to portfolio metadata.
- Sector: Ecosystem/platform building
- Application: Marketplace of agentic plugins and interoperable tools
- What it could deliver: A plug-in ecosystem where third parties contribute generators, predictors, and synthesis/assay modules with standard interfaces.
- Tools/products/workflows: Open APIs/SDKs, capability registries, benchmarking harnesses (using the MADD datasets), governance and security layers.
- Assumptions/dependencies: Community adoption; IP/licensing terms; security review for tool onboarding.
- Sector: Sustainability and operations
- Application: Resource-aware in silico-first discovery
- What it could deliver: Reduction in wet-lab waste and energy through smarter pre-filtering and compute scheduling; carbon-aware docking runs.
- Tools/products/workflows: Cost/carbon accounting integrated into orchestration; scheduling agents that balance accuracy, cost, and energy.
- Assumptions/dependencies: Telemetry from compute/wet-lab systems; organizational incentives for sustainability.
Notes on Feasibility, Risks, and Dependencies
- Data quality and availability: MADD’s performance depends on curated datasets linking molecules to target properties; DatasetBuilder can help, but gaps bias results.
- Compute and tooling: Docking and ML training require scalable compute and licenses/stack (RDKit, docking engines, LLM hosting).
- In silico vs. in vitro gap: Docking and IC50 predictions are proxies; wet-lab validation (assays, cells, in vivo) remains essential.
- Integration engineering: Current tool additions require code modifications; productionizing MADD needs MLOps, APIs, and audit logging.
- Governance and safety: Guardrails are needed to prevent misuse, ensure transparency (decision logs), and comply with regulatory and IP frameworks.
- Generalization: Strong results across tested cases, including an unseen thrombocytopenia dataset, but performance on entirely novel targets or modalities requires further study.
Glossary
Below is an alphabetical list of advanced domain-specific terms from the paper, each with a brief definition and a verbatim usage example from the text.
- ABL: A tyrosine kinase that serves as a biological target in drug discovery, often implicated in cancer. "STAT3, ABL, COMT, ACL, and PCSK9."
- ACL: ATP citrate lyase; a metabolic enzyme and drug target relevant to lipid metabolism and dyslipidemia. "STAT3, ABL, COMT, ACL, and PCSK9."
- AutoML: Automated machine learning; systems that automatically build, train, and select ML pipelines for specific tasks. "Furthermore, this toolkit features an AutoML framework FEDOT \cite{nikitin2022automated} designed to automatically train machine learning pipelines using custom datasets to predict molecular properties for new disease cases."
- AutoML-DL: An LLM-controlled tool in MADD that automates molecule generation, property prediction, and model training/monitoring. "The AutoML-DL tool is capable of generating molecules, predicting molecular properties, initiating training procedures, and monitoring the training status of both predictive and generative models."
- Binding affinity: A measure of the strength of interaction between a ligand (drug) and its target protein. "As a result, we identified several molecule hits with favorable bioactivity, binding affinity, and other physicochemical properties."
- BindingDB: A public database of measured binding affinities between small molecules and proteins. "It has access to the BindingDB and ChEMBL databases."
- Brenk: A medicinal chemistry structural filter that flags problematic or undesirable substructures in molecules. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- ChEMBL: A large bioactivity database of drug-like molecules used for model training and benchmarking. "It has access to the BindingDB and ChEMBL databases."
- COMT: Catechol-O-methyltransferase; an enzyme target associated with neurological conditions. "STAT3, ABL, COMT, ACL, and PCSK9."
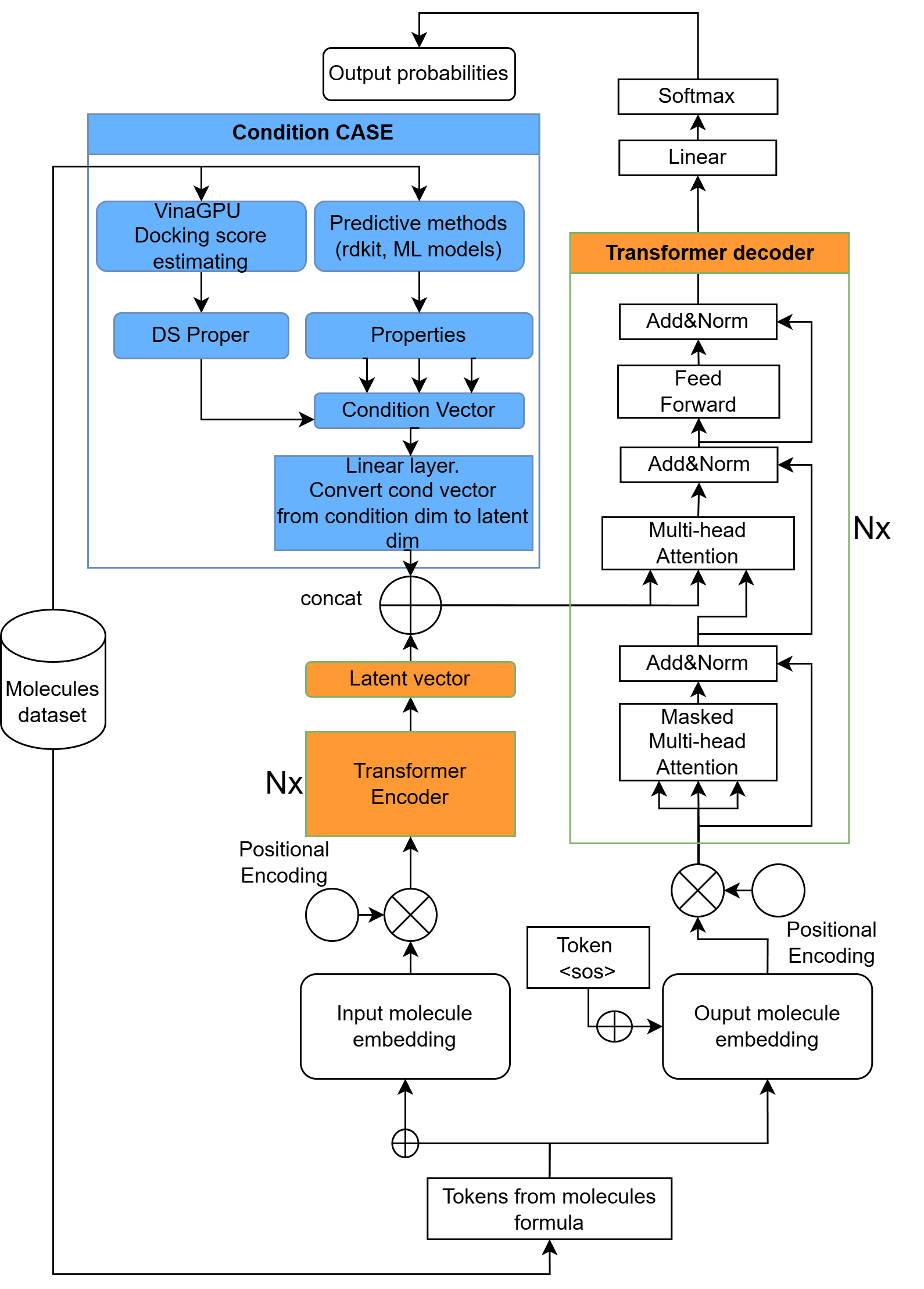
- CVAE: Conditional Variational Autoencoder; a generative model used to produce molecules with desired properties. "Currently, MADD supports two generative models: LSTM-based GAN and transformer-based CVAE."
- De novo: From scratch; generation or design of molecules without using existing templates. "MADD employs four coordinated agents to handle key subtasks in de novo compound generation and screening."
- Docking score: A computed estimate of binding energy from molecular docking, used to predict how well a ligand binds to a target protein. "This energy can be estimated using molecular docking, typically called the docking score."
- Drug-likeness (QED): Quantitative Estimate of Drug-likeness; a metric indicating how likely a molecule has properties consistent with successful drugs. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- FEDOT: An AutoML framework used to automatically create and train ML pipelines for molecular property prediction. "Furthermore, this toolkit features an AutoML framework FEDOT \cite{nikitin2022automated} designed to automatically train machine learning pipelines using custom datasets to predict molecular properties for new disease cases."
- Fragment-based RL (FBRL): A reinforcement learning approach that constructs molecules by assembling fragments to optimize target properties. "This study involved validation using one of the recently explored drug design cases on thrombocytopenia \cite{zavadskaya2025integrating}, where fragment-based RL was employed for the generation of novel SYK-inhibitors (hereafter, SYK-FBRL)."
- Glaxo: A structural filter set (originating from GlaxoSmithKline) used to flag undesirable substructures in molecules. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- GSK-3β: Glycogen synthase kinase 3 beta; a kinase targeted in Alzheimer’s and other diseases. "We conducted a case study on Alzheimer's disease to compare molecules generated by MADD with experimentally validated GSK-3 inhibitors from ChEMBL"
- IC50: Half-maximal inhibitory concentration; the concentration of a compound needed to inhibit a biological process by 50%. "MADD can use the tool to predict the half-maximal inhibitory concentration (IC50), which is the concentration of a drug required to inhibit a biological process or response by 50\% and is the most widely used and informative measure of a drug's efficacy."
- Kd: Dissociation constant; a measure of the affinity between a ligand and a protein, lower values indicate stronger binding. "Data acquisition requires specifying a target protein (or its respective database ID) and an affinity measurement type (Ki, Kd, or IC50)."
- Ki: Inhibition constant; describes the potency of an inhibitor binding to an enzyme or receptor. "Data acquisition requires specifying a target protein (or its respective database ID) and an affinity measurement type (Ki, Kd, or IC50)."
- Ligand: A molecule that binds to a target protein, often used to refer to drug candidates. "One of the target properties we used as training data was the binding energy of the target protein to a ligand."
- Monte Carlo tree search (MCTS): A probabilistic search strategy used to explore decision spaces, here applied to molecular generation. "a Monte Carlo tree search ChatChemTSv2 \cite{ishida2024large}"
- PAINS: Pan-Assay INterference compounds; filters identifying substructures that often produce false positives in assays. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- PCSK9: Proprotein convertase subtilisin/kexin type 9; a target involved in cholesterol regulation. "STAT3, ABL, COMT, ACL, and PCSK9."
- pIC50: The negative base-10 logarithm of IC50, used to linearize and stabilize potency values for modeling. "Using the AutoML framework, MADD could derive a pipeline to predict pIC50, achieving a metric value of R\textsuperscript{2}=0.75, compared to 0.78 in SYK-FBRL."
- RDKit: An open-source cheminformatics toolkit used for molecular operations and property calculations. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- SMILES: Simplified Molecular-Input Line-Entry System; a textual representation of molecular structures. "There are many strong generative models in drug design that are capable of producing molecules in SMILES notation."
- STAT3: Signal Transducer and Activator of Transcription 3; a transcription factor and drug target in oncology and inflammation. "STAT3, ABL, COMT, ACL, and PCSK9."
- SurehEMBL: A structural filter (related to SureChEMBL) used to flag undesirable substructures in molecules. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- SYK: Spleen tyrosine kinase; an immune-related kinase and drug target. "fragment-based RL was employed for the generation of novel SYK-inhibitors (hereafter, SYK-FBRL)."
- Synthetic accessibility (SA): A score estimating how easy it is to synthesize a molecule. "The Orchestrator can use several RDKit-based functions: synthetic accessibility (SA), drug-likeness estimation (QED), and structural filters such as Brenk, SurehEMBL, Glaxo, and PAINS."
- Tanimoto similarity: A fingerprint-based similarity metric between molecules, ranging from 0 (dissimilar) to 1 (identical). "Moreover, generated compounds demonstrated high structural diversity, with an average Tanimoto similarity of 0.43."
- Thrombocytopenia: A condition characterized by abnormally low platelet counts; used as a drug discovery case. "Thrombocytopenia case study."
Collections
Sign up for free to add this paper to one or more collections.