- The paper introduces SQMG that leverages chemistry-informed variational circuits and GPU-accelerated tensor-network simulation, achieving linear qubit scaling.
- It demonstrates significant speedups, with GPU state-vector and tensor-network simulations outperforming CPU baselines and enabling simulation up to 40 heavy atoms.
- Bayesian optimization outperforms COBYLA in tuning quantum circuit parameters, enhancing the efficiency of molecular generation tasks.
Scalable Quantum Molecular Generation via GPU-Accelerated Tensor-Network Simulation
Motivation and Context
Efficient exploration of chemical space is foundational for advances in drug discovery, materials science, and chemical synthesis. Prevailing generative models—GANs, VAEs, autoregressive flows, and LLMs—have demonstrated substantial capability in molecular graph generation, but are constrained by high parameter counts, substantial training data requirements, and susceptibility to mode collapse and limited controllability. Quantum generative models, leveraging variational quantum circuits, present an alternative approach: parameterized quantum circuits define nonlinear sampling distributions over exponentially large Hilbert spaces, enabling direct sampling via measurement. However, dynamic circuit control and classical simulation bottlenecks have limited the scalability of prior frameworks, notably the dynamic QMG approach. The work introduces Scalable Quantum Molecular Generation (SQMG), which integrates chemistry-informed circuit design with GPU-accelerated tensor-network simulation, offering a scalable and reproducible testbed for quantum molecular generation.
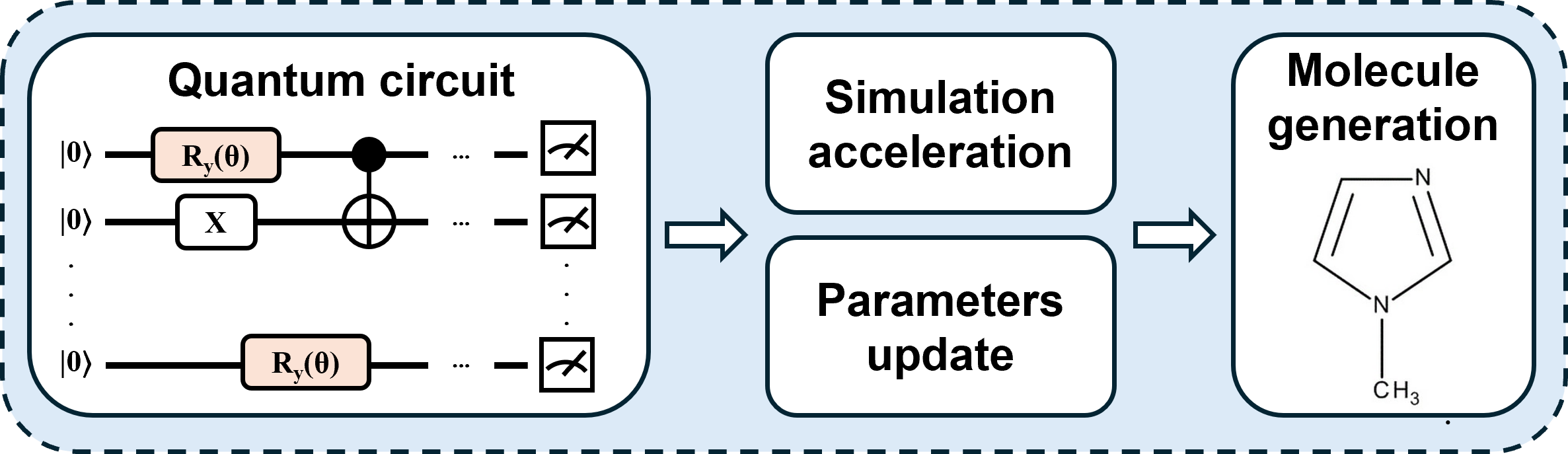
Figure 1: Workflow of quantum molecular generation, from quantum circuit initialization through iterative sampling and classical decoding.
SQMG Circuit Architecture
SQMG employs a hybrid, chemistry-guided variational ansatz with an "atom no-reuse, bond reuse" paradigm, assigning fixed three-qubit registers to each heavy atom and sequentially reusing a two-qubit bond register for bond generation. For a molecule with N heavy atoms, the circuit utilizes $3N+2$ qubits—linear scaling with respect to system size, in contrast to quadratic growth in static circuit designs. Atomic registers encode element identity (seven heavy atom types + NONE) via three-bit classical codes, while the bond register encodes bond order across four states (no bond, single, double, triple). The circuit structure eliminates frequent mid-circuit resets and classical conditionals required by atom-reuse designs, thus minimizing circuit depth and synchronization overhead, optimizing high-throughput sampling.
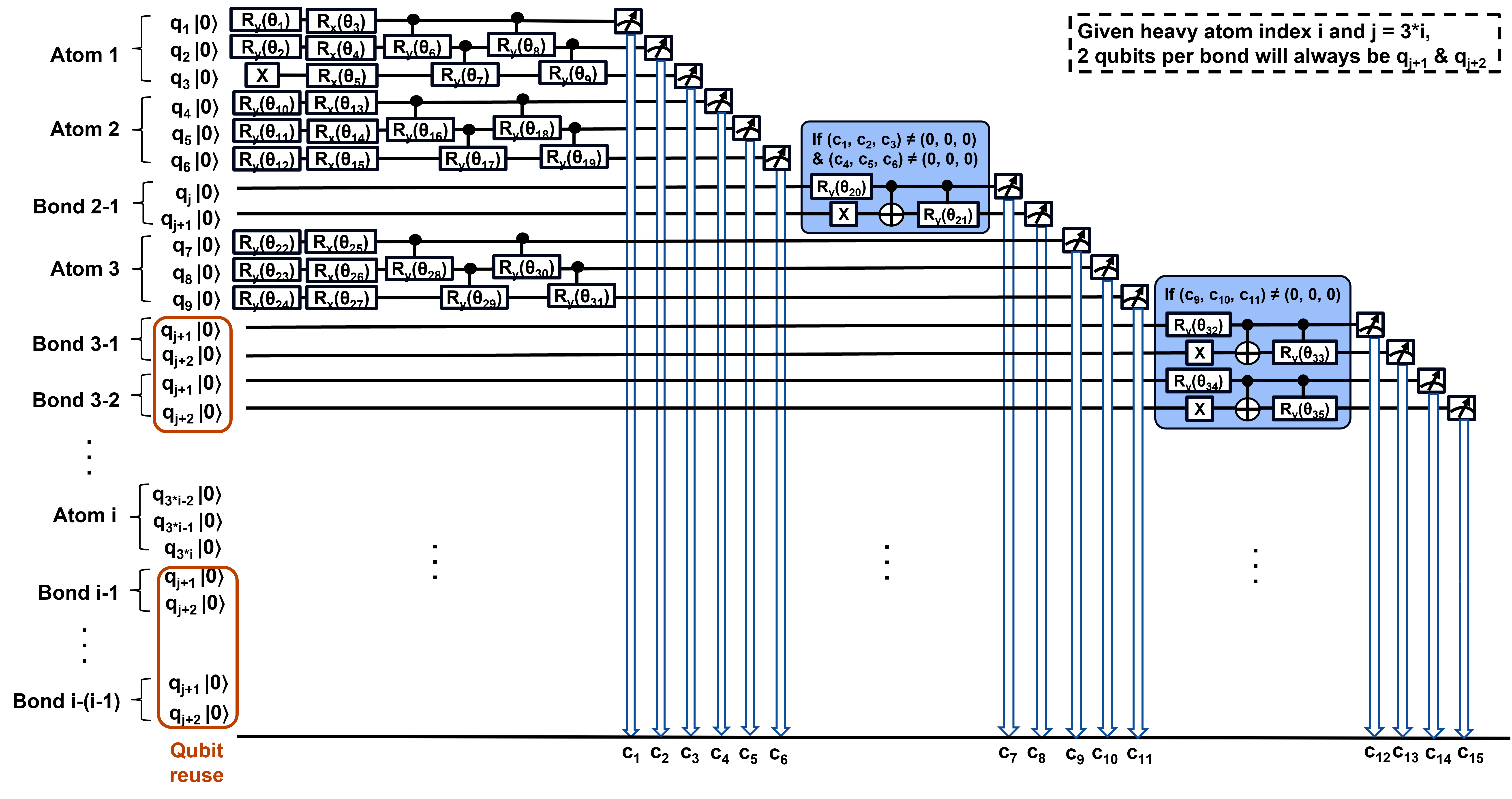
Figure 2: Quantum circuit diagram underpinning SQMG with dedicated atomic qubits and sequential bond-qubit reuse, ensuring linear qubit scaling.
The architecture's static atomic registers streamline tensor network contraction, facilitating scalable simulation on classical hardware for larger systems. The parameter scaling (N2+9N−1) provides sufficient expressivity for diverse molecular topologies while maintaining architectural regularity.
GPU-Accelerated Simulation Backends
SQMG is implemented in CUDA-Q and benchmarked across three backends: state-vector simulation on CPU, state-vector simulation on GPU (cuStateVec), and tensor-network simulation on GPU (cuTensorNet). State-vector simulation is optimal for small-to-medium systems, where GPU parallelism yields pronounced speedups. However, exponential storage (23N+2) renders state-vector approaches infeasible as N increases, even on high-memory GPUs. Tensor-network simulation contracts structured tensor graphs instead of explicit state-vectors, mitigating memory bottlenecks and enabling exact simulation for up to N=40 heavy atoms.
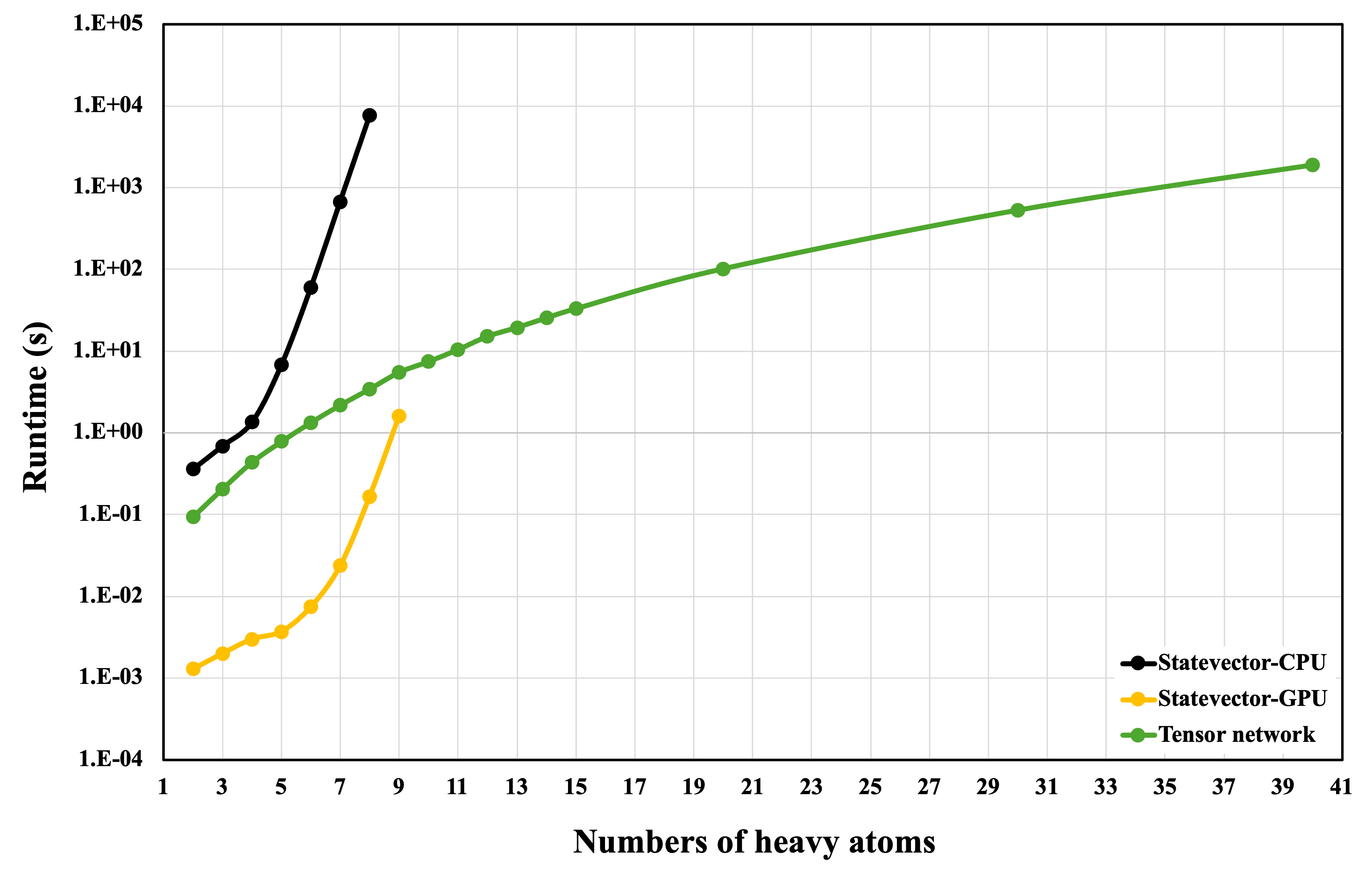
Figure 3: Runtime scaling of CUDA-Q simulation backends, highlighting the superior scalability of GPU-accelerated tensor-network contraction for large circuits.
Key numerical results:
- At N=8, state-vector simulation (GPU) achieves a 4.5×104 speedup over CPU state-vector simulation, with tensor-network (GPU) yielding a 2.2×103 speedup relative to the CPU baseline.
- Tensor-network backend remains computationally tractable at N=40, where all state-vector approaches fail due to memory overflow.
The authors evaluate runtime scaling for both atom no-reuse and atom reuse circuit configurations under tensor-network simulation. For small $3N+2$0, runtime differences are negligible; as $3N+2$1 increases, static atomic registers significantly improve contraction efficiency. At $3N+2$2, the static version outperforms atom-reuse by $3N+2$3 despite increased qubit count, underscoring the advantage of structural regularity for scalable tensor-network contraction.
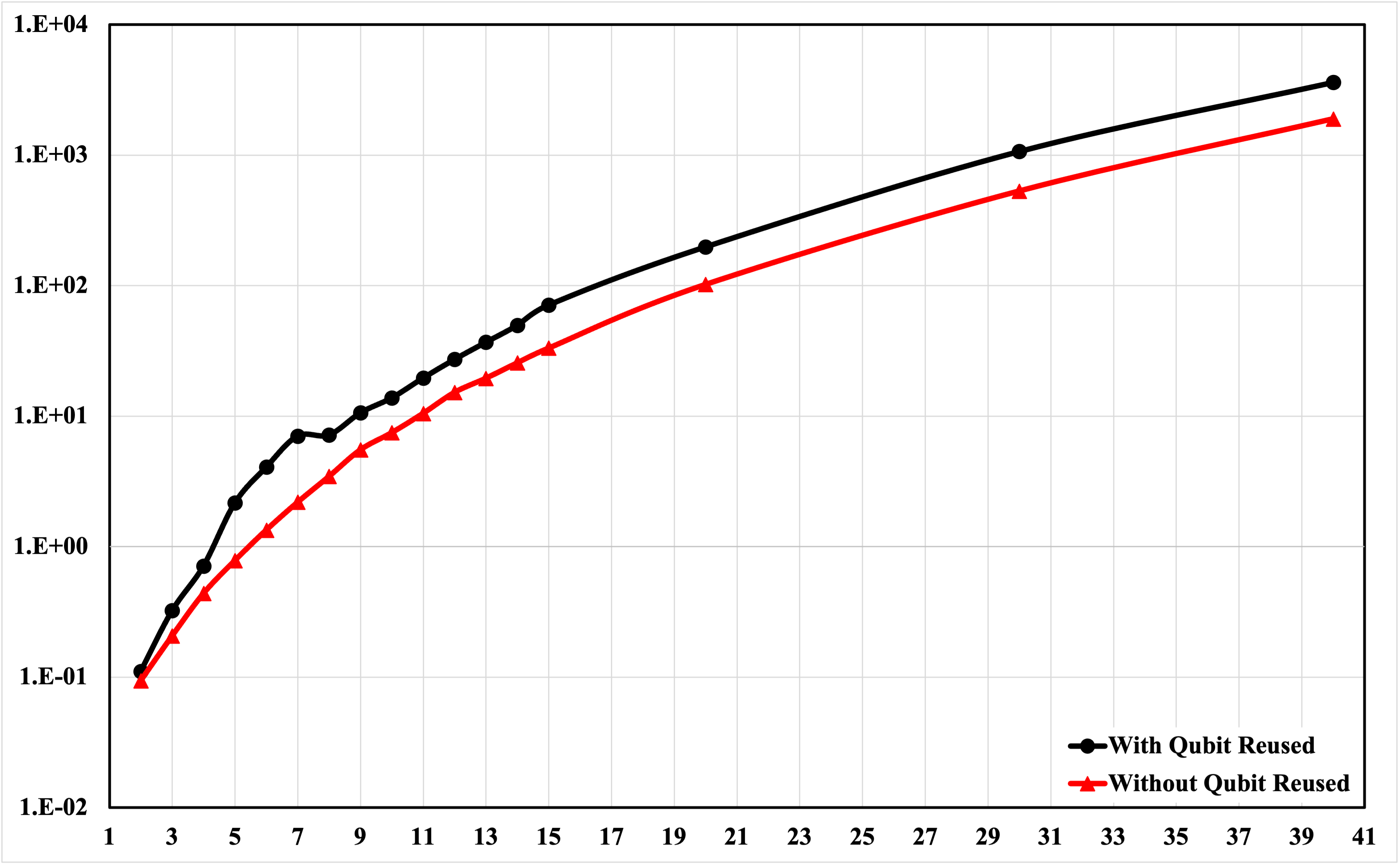
Figure 4: Runtime comparison for circuits with and without atom qubit reuse reveals marked performance advantages for static atomic registers under tensor-network simulation.
Training: COBYLA vs Bayesian Optimization
Training is benchmarked using composite objectives (Validity$3N+2$4Uniqueness) with two optimization strategies: COBYLA (derivative-free, trust-region) and Bayesian Optimization (BO) leveraging Gaussian Process surrogates and Expected Improvement acquisition. COBYLA converges rapidly but saturates at suboptimal values (0.32 Validity$3N+2$5Uniqueness), while BO maintains higher variance due to exploration, ultimately achieving superior optima (0.69 Validity$3N+2$6Uniqueness). This demonstrates BO's advantage in noisy, multimodal search landscapes typical of quantum circuit parameterization.
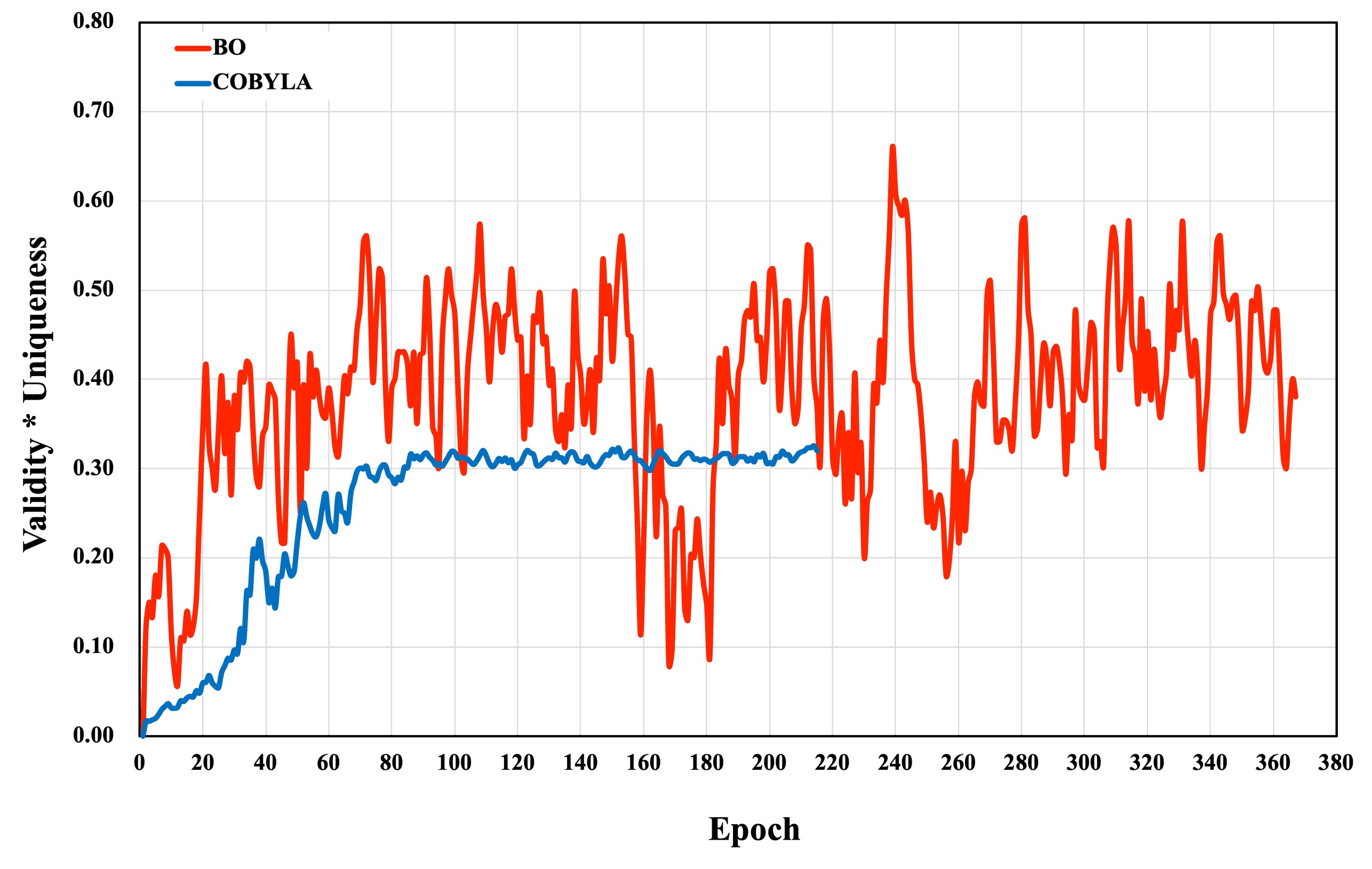
Figure 5: Optimization trajectories showing rapid but locally-constrained convergence of COBYLA and superior exploration by Bayesian Optimization, with higher final composite scores.
Molecular Generation Functionalities
SQMG supports three primary modes: de novo generation (full molecule synthesis), scaffold decoration (substituent selection with fixed core), and linker generation (connecting terminal fragments via quantum-generated linker algorithms).
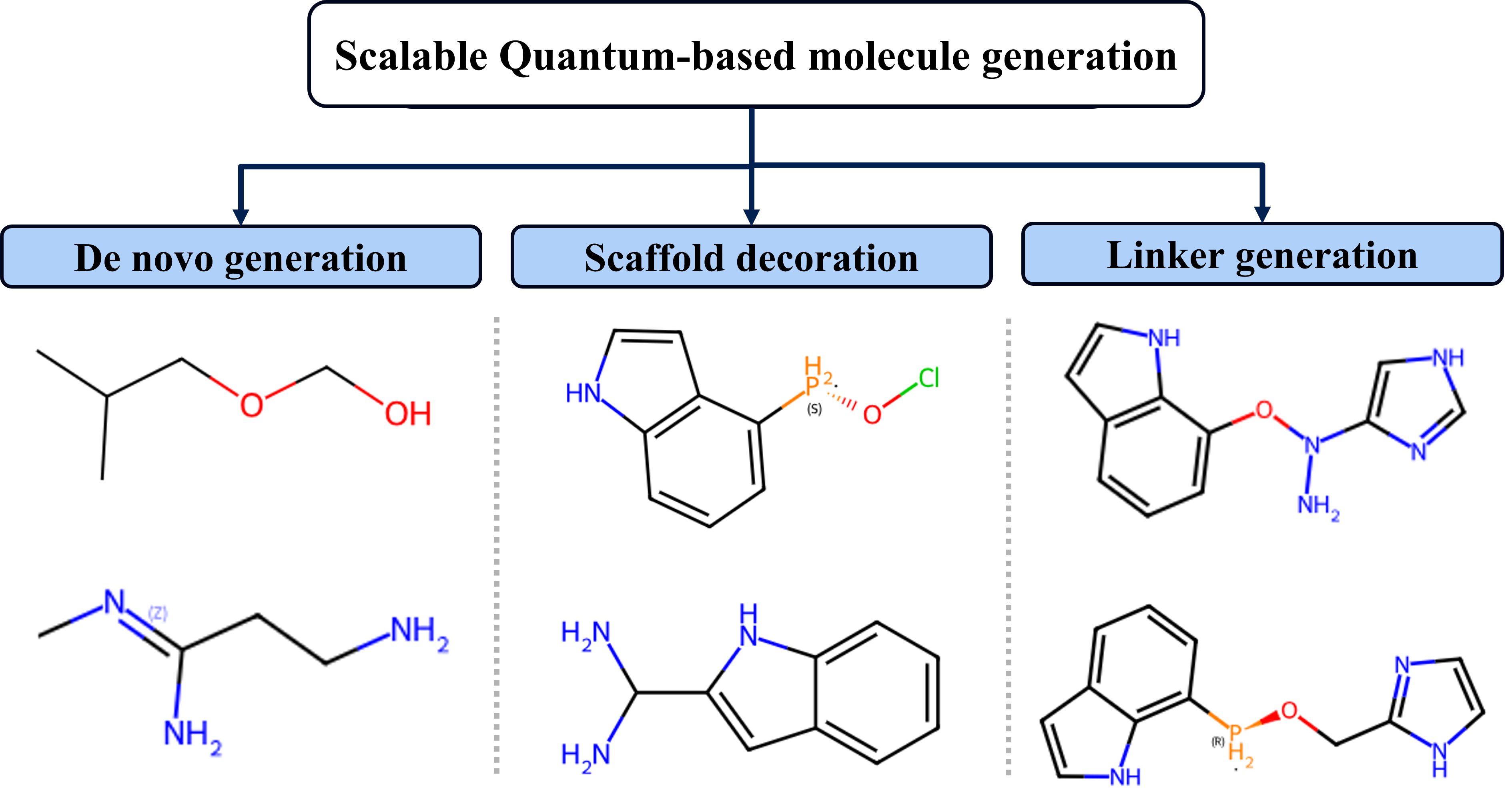
Figure 6: SQMG functionalities: full molecule de novo construction, scaffold decoration with variable substituents, and flexible linker generation bridging fragments.
Such versatility enables direct application to molecular design tasks ranging from pharmacophore expansion to materials assembly, while preserving the reproducibility and controllability inherent to quantum circuit sampling.
Implications and Future Directions
The integration of chemistry-inspired circuit design with GPU-accelerated tensor-network simulation enables linear qubit scaling and tractable simulation of circuits with up to $3N+2$7 heavy atoms—substantially extending previous boundaries that were constrained by state-vector memory limitations. The empirical advantage of static atomic registers in circuit structure highlights the importance of architectural regularity for simulation efficiency in tensor-network backends. Bayesian optimization's efficacy for quantum circuit parameter search suggests further exploration of advanced surrogate-based optimizers, possibly with adaptive objective functions for generative model calibration.
Practically, SQMG establishes a reproducible platform for benchmarking future quantum generative molecular algorithms, particularly as quantum hardware matures toward intermediate-scale devices. The framework can be readily extended to include additional atomic/bond states, multi-objective optimization, and integration with classical cheminformatics.
Conclusion
Scalable Quantum Molecular Generation (SQMG) offers a chemistry-guided variational circuit architecture coupled with GPU-accelerated tensor-network simulation, achieving linear qubit scaling and robust simulation for large molecules. SQMG provides strong numerical speedups and supports versatile generation modes, with Bayesian optimization outperforming local search in parameter tuning. The work establishes a scalable, structured, and reproducible benchmark for quantum molecular generation, advancing both practical simulation methodologies and theoretical understanding of quantum generative models in molecular design contexts.