- The paper shows that tabular in-context learners, TabPFN3 and TabICL, outperform classical baselines and fine-tuned models in protein fitness prediction tasks.
- It demonstrates strong few-shot learning and sample efficiency across ProteinGym and PpEST, highlighting rapid performance gains with small support sets.
- The study reveals that representation quality predominantly drives performance in small-molecule property prediction and out-of-distribution generalization.
Tabular In-Context Learning for Biomolecular Property Prediction: An Empirical Evaluation
Introduction
This paper systematically examines the generalization capabilities of tabular in-context learners (ICLs)—specifically TabPFN3 and TabICL—on biomolecular property prediction tasks. The focus is twofold: regression of protein fitness based on fixed sequence embeddings, and classification of small-molecule properties using descriptor-based representations. A central hypothesis is that tabular ICLs, pretrained on synthetic data generated from random causal graphs, can be effective predictors in biological domains, even without explicit structural or domain knowledge. The study evaluates these models in both protein engineering (ProteinGym, PpEST) and chemical informatics (TDC ADMET, MoleculeNet, FS-Mol, DrugOOD), comparing their performance against standard supervised baselines and task-specific models.
Tabular In-Context Learners and Biomolecular Representations
Tabular foundation models operate by encoding in-context support sets as tables, then performing amortized Bayesian inference via transformer architectures without task-specific fine-tuning. The two main models, TabPFN3 (with regression and scalability enhancements) and TabICL (with column/row attention compression), are evaluated here as predictors paired with fixed representations.
Protein sequences are embedded using ESMC (a 300M-parameter protein LLM), yielding 960-dimensional vectors as inputs to all models. For small molecules, descriptors include ECFP/Morgan fingerprints, RDKit physicochemical features, and their concatenations. These are compared with graph-based models (ChemProp, ChemProp+CheMeleon) operating directly on molecular structure.
Protein Fitness Prediction: Methodology and Results
Experimental Setup
ProteinGym (217 deep mutational scanning assays) and PpEST (esterase family dataset) serve as benchmarks. All models operate under fixed ESMC embeddings, tested under full-training and few-shot regimes. Performance metrics are Spearman rank correlation and MSE. Baselines include ridge regression, gradient boosting, and fine-tuned ESMC.
TabPFN3 and TabICL outperform classical baselines and fine-tuned ESMC in aggregate metrics. Under random 5-fold cross-validation:
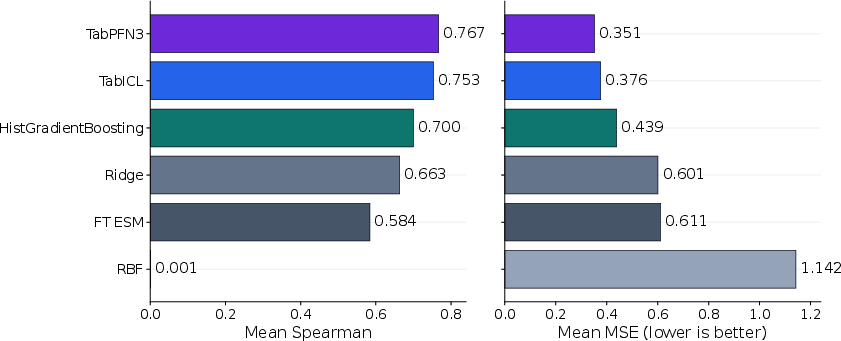
Figure 1: ProteinGym random 5-fold method comparison across 217 assays; TabPFN3 is strongest, TabICL remains close, and both surpass classical baselines.
Performance differences between TabPFN3 and TabICL are broad but modest across individual assays.
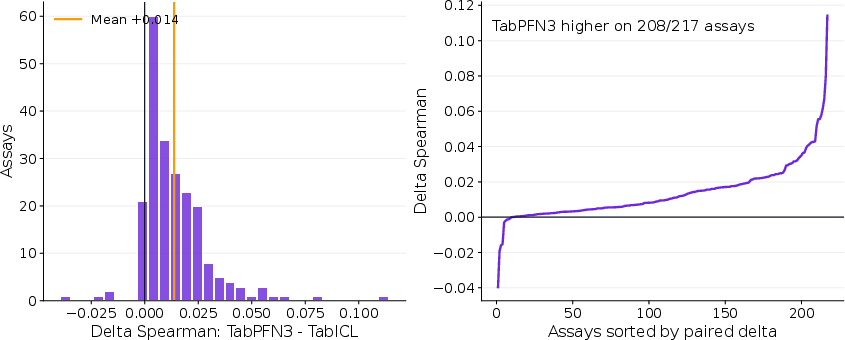
Figure 2: Assay-level paired difference in validation performance between TabPFN3 and TabICL on ProteinGym. Positive values indicate higher Spearman correlation for TabPFN3.
On official holdout splits (random, modulo, contiguous), TabPFN3 consistently outperforms TabICL, though both drop substantially on harder splits.
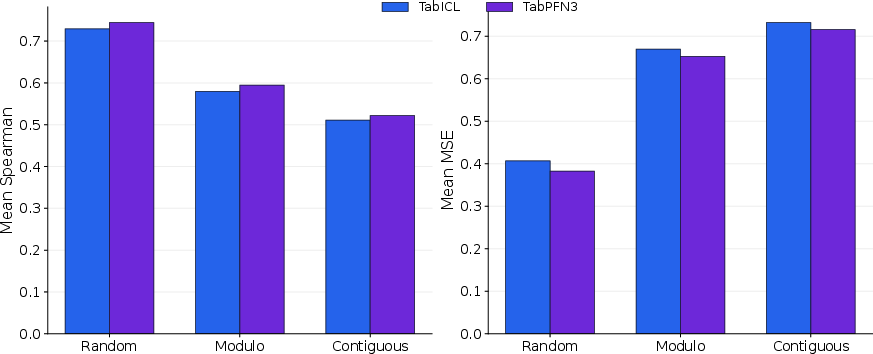
Figure 3: ProteinGym performance across random, modulo, and contiguous holdout schemes; TabPFN3 modestly exceeds TabICL, but absolute performance declines on harder splits.
Few-shot learning curves demonstrate strong sample efficiency, especially with TabPFN3 in the low-label regime.
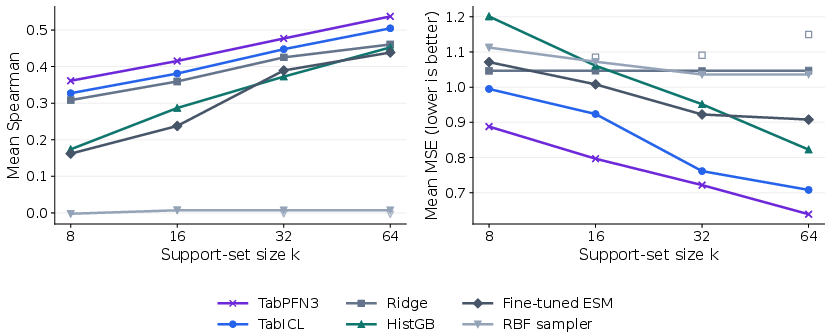
Figure 4: ProteinGym few-shot performance as support-set size increases; TabPFN3 and TabICL adapt rapidly with increasing support.
Assay size does not fully explain variability in model performance, highlighting the role of representation geometry.
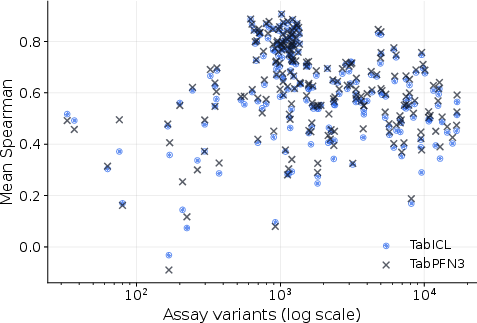
Figure 5: ProteinGym assay-size diagnostic; mean Spearman plotted against assay variant count for TabPFN3 and TabICL.
PpEST Results
Both TabICL and TabPFN3 deliver superior performance across diverse esterase endpoints, trading off minor differences in MSE and Spearman correlation.
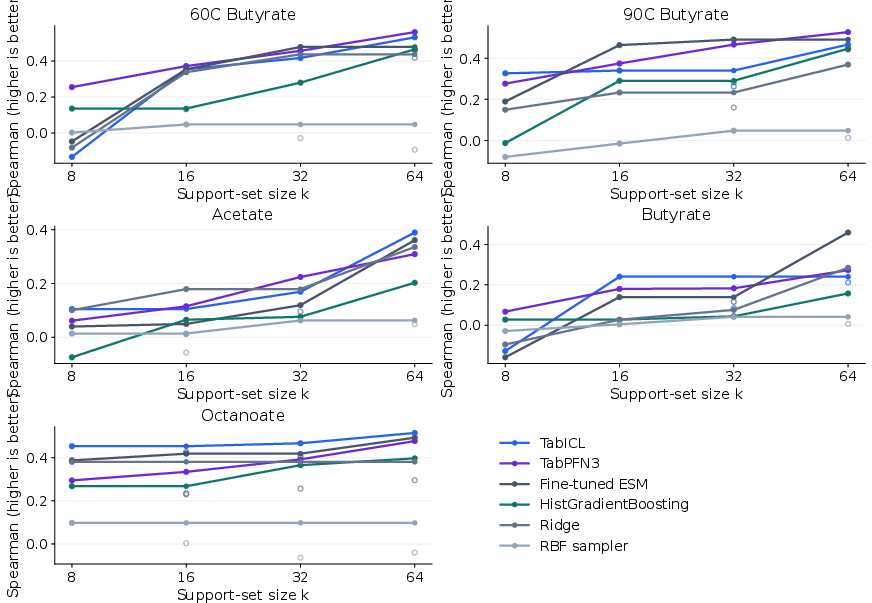
Figure 6: Few-shot PpEST performance measured by Spearman correlation; TabICL and TabPFN3 exhibit strong rank preservation with limited samples.
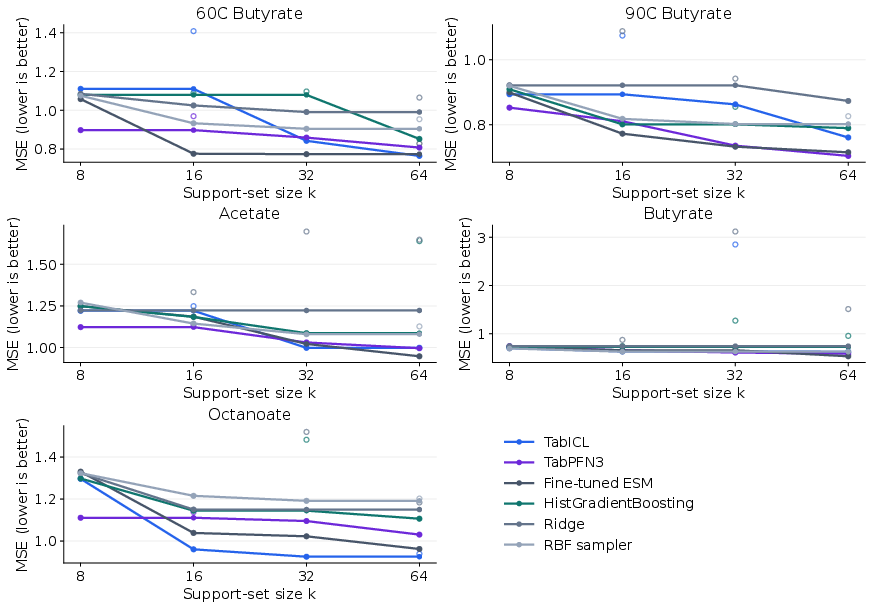
Figure 7: Few-shot PpEST performance measured by MSE; both models improve consistently as support-set size increases.
Small-Molecule Property Prediction: Methodology and Results
Experimental Setup
Four benchmark families are considered: TDC ADMET, MoleculeNet, FS-Mol, DrugOOD. Models are evaluated as descriptor--predictor pairs, with task-level aggregation.
TabPFN3 and TabICL are competitive in few-shot molecular property classification, though rankings depend heavily on descriptor choice and benchmark family.
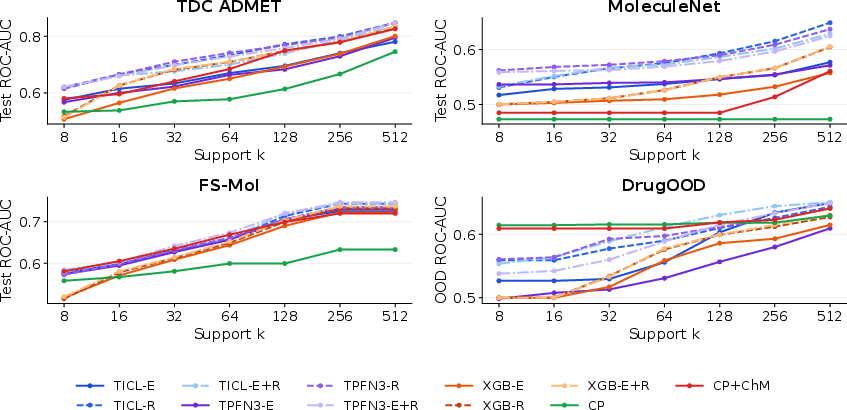
Figure 8: Few-shot small-molecule learning curves; best-so-far task-averaged ROC-AUC trends across benchmarks.
In the full-training regime, TabPFN3 and TabICL attain strong performance, with distinct winner pairs depending on benchmark definition.
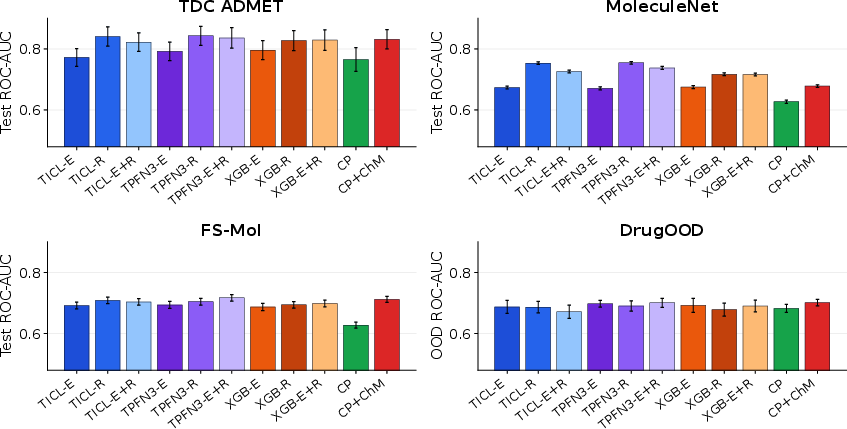
Figure 9: Full-train small-molecule benchmark summary; bars show mean ROC-AUC across tasks for different model-representation pairings.
Out-of-Distribution Generalization
DrugOOD analysis reveals nontrivial ID/OOD generalization gaps, which are influenced by model architecture and molecular representation.
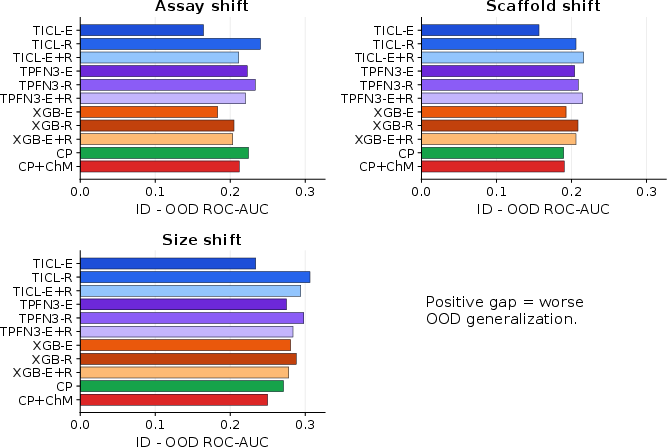
Figure 10: DrugOOD full-train ID/OOD generalization gap; larger positive values indicate greater performance drop under OOD shift.
Paired comparisons show TabPFN3 and TabICL are closely matched, with marginal gains varying by task and representation.
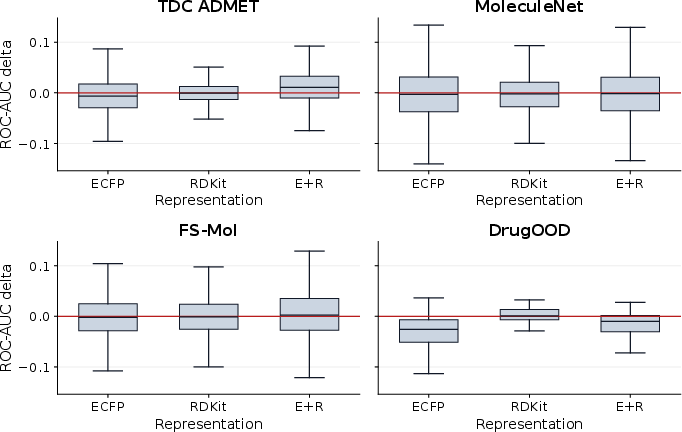
Figure 11: Paired few-shot comparison of TabPFN3 and TabICL on molecules; positive values denote higher ROC-AUC for TabPFN3.
Discussion
Empirically, tabular ICLs are robust predictors for protein fitness regression and competitive in molecular classification, provided the underlying representation exposes sufficient structural information. This demonstrates that amortized pretraining on synthetic causal graphs can transfer effectively to biological domains, so long as molecular or sequence features serve as suitable "tabularized" proxies for structure and function. However, there is no universal dominance: representation choice remains a primary determinant of outcome, especially in chemical benchmarks. Official holdout protocols (ProteinGym modulo/contiguous, DrugOOD OOD) underscore the importance of rigorous evaluation for claims of generalization.
Theoretically, this pattern is consonant with ICL's interpolation regime, where geometric locality in embedding space enables label extrapolation from few support examples. Practically, it reinforces the utility of pairing strong pretrained encoders (ESMC, ECFP/RDKit) with tabular ICLs as plug-and-play predictors across biomolecular domains. Limitations include dependence on representation quality, variability in out-of-distribution robustness, and operational constraints for large-scale benchmarks.
Implications and Future Directions
From an applied perspective, the findings validate TabPFN3 and TabICL as viable options for few-shot biomolecular screening, potentially accelerating wet-lab cycles in protein engineering and drug discovery. The separation of representation and predictor effects motivates future work toward jointly optimized paired models, adaptive feature selection, and domain-conditional pretraining. Furthermore, extending tabular ICLs to accommodate graph-based molecular encodings or hybrid strategies may improve OOD performance in chemical informatics.
Mechanistically, deeper analysis of the relationship between synthetic causal priors and real biological data would clarify transfer regimes and may unlock further improvements in sample efficiency. Moreover, systematic audits under rigorous distributional shifts should remain central to claims of robustness.
Conclusion
Tabular foundation models such as TabPFN3 and TabICL are competitive predictors for biomolecular property tasks, achieving strong performance when coupled with fixed biological or chemical representations. Their efficacy is shaped more by the quality and structure of these representations than by in-context adaptation alone. While generalization to protein and small-molecule benchmarks is empirically supported, careful protocol design and separation of learner--representation effects are essential for credible scientific auditing.