- The paper introduces a novel chemically-grounded benchmark suite and RL post-training method to evaluate and optimize LLM performance in drug design.
- It benchmarks open and closed LLMs across property prediction, representation transformation, and generative tasks, revealing significant gains and persistent performance gaps.
- Findings show that RL post-training boosts constrained molecule generation while highlighting challenges in experimental property prediction and complex multi-turn optimization.
Evaluation of LLM Progression in Small-Molecule Drug Design
Introduction
This paper systematically analyzes the progression of LLM capabilities in small-molecule drug design, leveraging a novel suite of chemically-grounded benchmarks spanning molecular property prediction, representation transformation, and generative design tasks. The core contribution is twofold: (1) formalizing a diverse set of real-world chemistry tasks as RL environments to enable targeted evaluation and domain sharpening via RL-based post-training, and (2) benchmarking both open-weight and closed frontier LLMs across single- and multi-turn molecular discovery settings to elucidate practical capability gaps and potential for improvement through post-training.
The task suite encompasses six major classes: RDKit-derived property prediction, experimental property (potency and DMPK) regression and classification, molecular representation transformation, multiproperty-constrained molecule generation, multiple-choice graph/representation discrimination, and assorted tasks such as substructure classification and reaction outcome prediction. Each is posed as an RL environment with dense or sparse reward, tailored to effectively probe a range of competencies from functional group recognition to chemical nomenclature parsing and generalization across chemical space.
The RL post-training protocol builds on the DAPO variant of Group Relative Policy Optimization (GRPO), which standardizes reward advantages within sampled groups to produce computationally efficient, robust policy improvement. The approach eschews value-function approximation, relying instead on large group sampling to stabilize policy gradients and leverage dense, structured reward signals defined for each chemical task.
RL-Based Post-Training: Qwen3-30B to Aspen
A principal experimental axis is the RL post-training of the open Mixture-of-Experts model Qwen3-30B-A3B-Thinking-2507. Training is carried out over ∼900k chemically-diverse prompts and 30B parameters, using a single-epoch, highly parallelized GRPO procedure. Reward curves demonstrate steady and often sigmoidal improvements, notably in the constrained generation and certain transformation tasks, but also reveal clear bottlenecks in experimental property prediction tasks—especially in low-data or highly OOD (out-of-distribution) regimes.
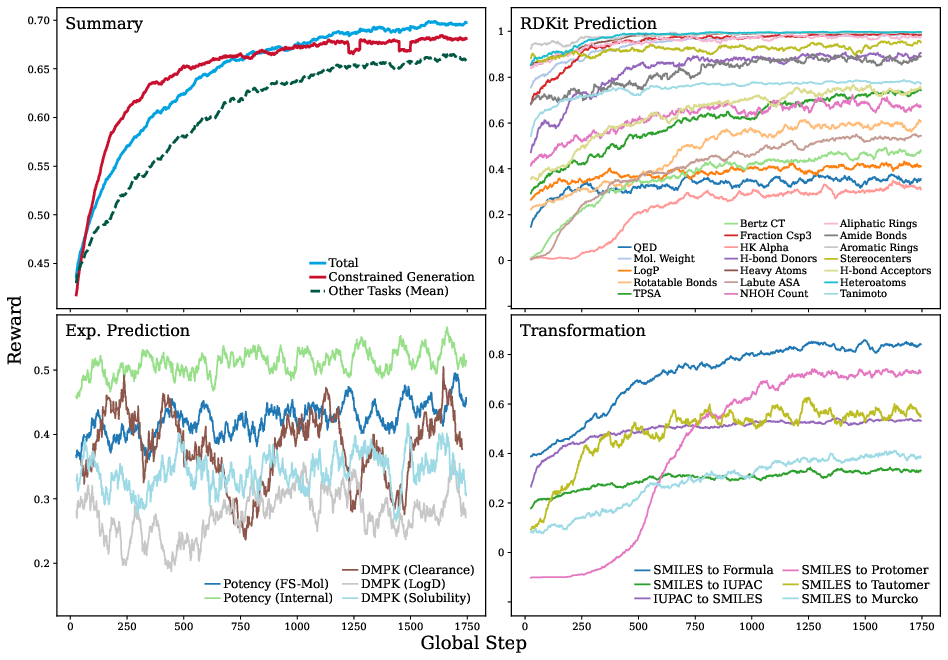
Figure 1: Reward trajectories during RL post-training, with pronounced and rapid gains for constrained generation tasks but more stunted progress in data-sparse experimental settings.
The general finding is that RL can “sharpen” latent chemical knowledge in the base LLM, yielding rapid improvements where prior competence exists, but is fundamentally limited in OOD or knowledge-poor regimes, where RL reward shaping alone cannot compensate for absent foundational knowledge.
Single-Turn Model Capability Benchmarking
Benchmarking across multiple LLM families (Qwen/Aspen, OpenAI GPT-5/5.2, Claude Opus 4.0/4.6) reveals clear, interpretable patterns in competence progression.
RDKit-derived properties: Simple, mostly compositional properties (e.g., atom counts, fraction sp3) are increasingly robust across all families, but derived and context-dependent descriptors (e.g., H-bond donors, TPSA, QED, rotatable bonds) remain challenging—requiring both explicit chemistry knowledge and non-trivial graph reasoning. Aspen’s post-training yields strong gains (e.g., H-bond donors jump to R2=0.80), but local-valued, context-sensitive counts such as NH/OH remain problematic, implicating incomplete semantic understanding in current models.
Experimental property prediction and multiple-choice: Both regression and classification over sparse, in-context experimental data (e.g., SAR, DMPK, FS-Mol) separate the strongest closed models from others; performance remains low and non-monotonic even post-training, with all models failing to solve DMPK solubility prediction (all R2<0). This deficiency underlines the need for broader domain pre-exposure and more chemically focused midtraining routines.
Molecular transformations: Representation translation accuracy (e.g., SMILES↔IUPAC, protomer/tautomer generation) remains a difficult test case for all but the very largest closed models. Aspen demonstrates partial regularity learning (dense reward helps), yet only closed models (notably Opus 4.6) approach high accuracy for conversion between languages (IUPAC→SMILES, ∼0.55 accuracy).
Multiproperty-constrained generation: A practical highlight is Aspen’s strong improvement in constraint satisfaction and validity rates (from 0.09 to 0.21 for all-constraint satisfaction post-training), matching or exceeding several closed models and demonstrating that RL can close substantial capability gaps in design-centric settings.
Higher-order reasoning and reaction outcomes: Routine tasks, such as scaffold retention and substructure classification, are now robust, but tasks demanding explicit multi-graph reasoning (e.g., reaction outcome prediction, MCS finding) show persistent limitations. Improvement comes from both RL post-training (for Qwen/Aspen) and, more markedly, from Anthropic’s incorporation of chemical reasoning in newer models.
Multi-Turn Simulated Lead Optimization
The paper advances beyond single-turn evaluation into a simulated iterative design loop: docking score optimization for a real protein target under realistic DMPK/physicochemical constraints.
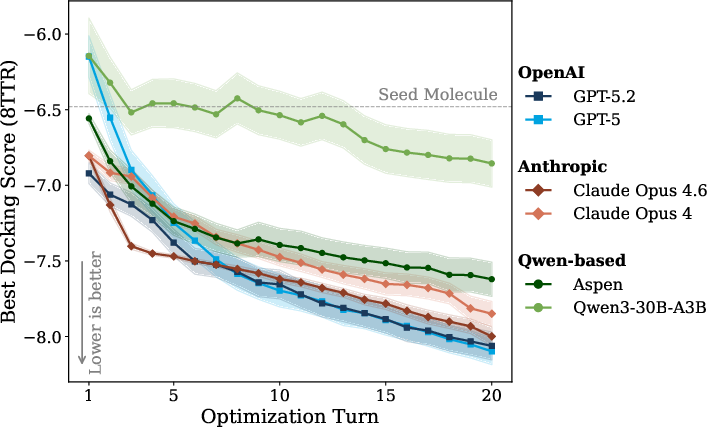
Figure 3: Multi-turn mean best docking score over 20 optimization steps; later model versions demonstrate superior optimization efficiency and better final scores, especially Aspen compared to baseline Qwen.
Optimization efficiency and validity: All families show improvement in chemical optimization with newer model iterations. Aspen, post RL-training, outperforms its base model both in finding low docking-score molecules and maintaining high validity/scaffold retention rates (see also Figures 6 and 7).
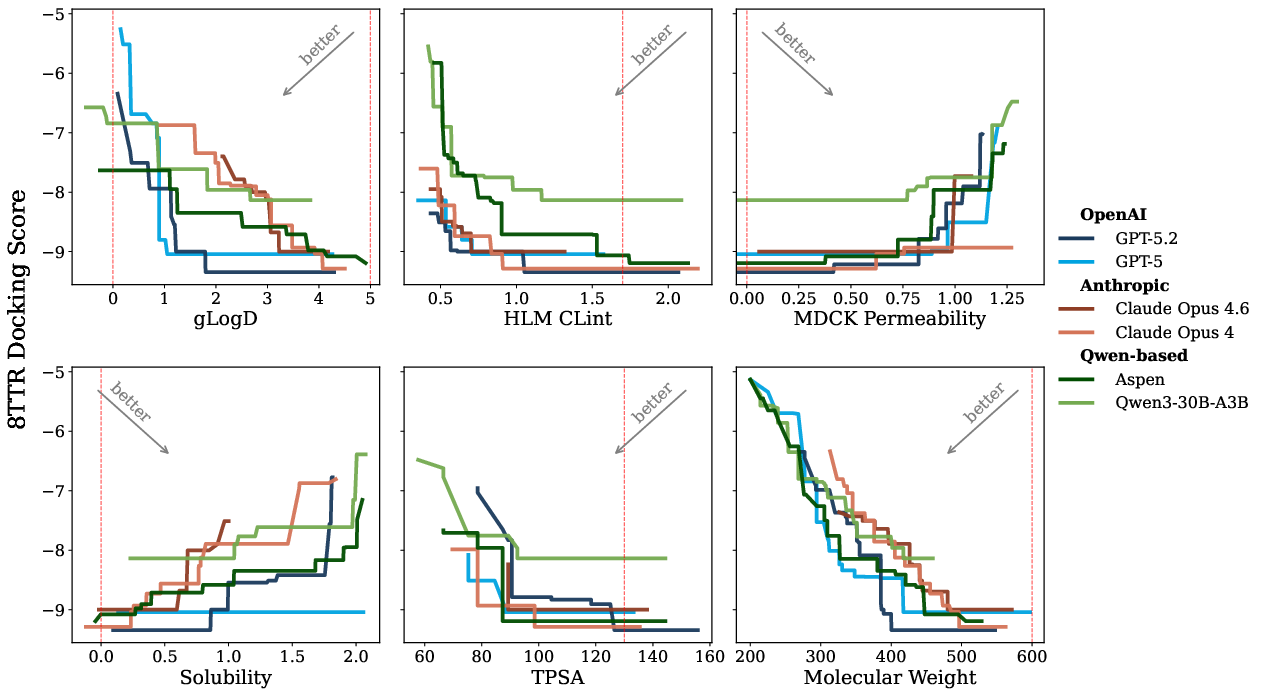
Figure 2: Pareto trade-off between docking score and key constraints, clarifying each model’s ability to balance potency improvement and property compliance; Aspen is more ligand-efficient, achieving better scores without size inflation.
Constraint satisfaction: Detailed analysis of constraint adherence (liver clearance, solubility, permeability, MW, TPSA, scaffold retention) finds that Qwen3 base and Aspen struggle to maintain HLM CLint compliance, trending toward “potency at any cost” as optimization progresses, whereas closed models maintain stronger constraint satisfaction overall (see Figure 5).
Chemical strategy and diversity: Analysis of molecular modification strategies (Figure 6) reveals that closed models exploit established medicinal chemistry heuristics (fluorination, preferred linker modifications), while Aspen learns a competitive, but narrower, set of productive transformations. Notably, Opus 4.6 suffers from reduced chemical diversity, suggesting emerging mode collapse during its training regime (Figure 7).
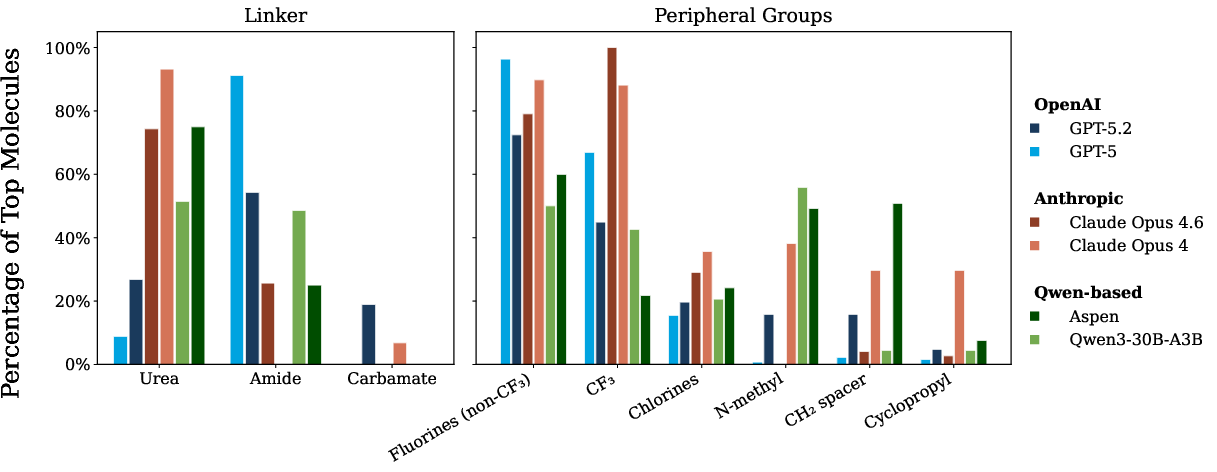
Figure 4: Chemical strategies employed by top-performing molecules; closed models (Opus 4.6) favor different linker and substitution patterns compared to Aspen and Qwen, reflecting richer domain priors.
Practical and Theoretical Implications
This work demonstrates that RL-based post-training can substantially elevate domain-specific performance of open models, in some generative tasks nearly closing the gap to contemporary closed “frontier” LLMs. However, post-training is maximally effective only where baseline domain knowledge is non-trivial—highlighting the continuing importance of base model midtraining on chemistry-rich corpora and the value of multi-modal, reward-shaped fine-tuning. A clear implication is that synthetic LLM chemical agents will require both base and targeted post-training, leveraging proprietary or task-tailored internal data.
Theoretically, the divergent patterns in task proficiency (“jagged frontier”) reinforce that LLM scaling and task exposure yield uneven gains, and that compositional chemical reasoning and inter-representation translation are among the most persistent weaknesses in current architectures. Subsequent model iterations (especially in the Anthropic family) evidence more deliberate, non-monotonic gains in chemically meaningful reasoning, likely due to focused training on such capabilities.
Practically, the results identify drug discovery tasks where LLMs can already contribute productively (e.g., multiproperty molecular generation, representation transformation under certain constraints), while underscoring persistent gaps in experimental data generalization and complex mechanistic chemical reasoning. Closing these gaps will necessitate deliberate curriculum design, chemistry-focused midtraining, and the integration of experimental knowledge unavailable in public datasets.
Future Directions
Several promising avenues are presented for the advancement of molecular LLMs:
- Joint scaling of architecture, chemistry-aware pretraining, and RL/SFT routines.
- Expansion of the environment/task repertoire to more faithfully reflect agentic workflows and decision processes in medicinal chemistry.
- Incorporation of proprietary measured data and advanced oracles, particularly for DMPK and off-target profiles.
- Focus on diversity retention and trade-off navigation (to prevent mode collapse) in generative chemical design.
- Rigorous ablation of training components to isolate sources of knowledge acquisition and generalization in chemical subdomains.
Conclusion
This paper provides a detailed, evidence-based framework for benchmarking and advancing LLM capabilities in small-molecule drug design, making significant strides in task formulation, environment construction, and RL-based model tuning. Strong empirical results support the assertion that RL post-training enables substantial progress for open models where domain knowledge is adequate, and elucidates the limits of current approaches in truly OOD or data-poor experimental regimes. These insights inform a research agenda toward LLMs robust enough to act as dependable computational agents in early-phase medicinal chemistry and beyond.

