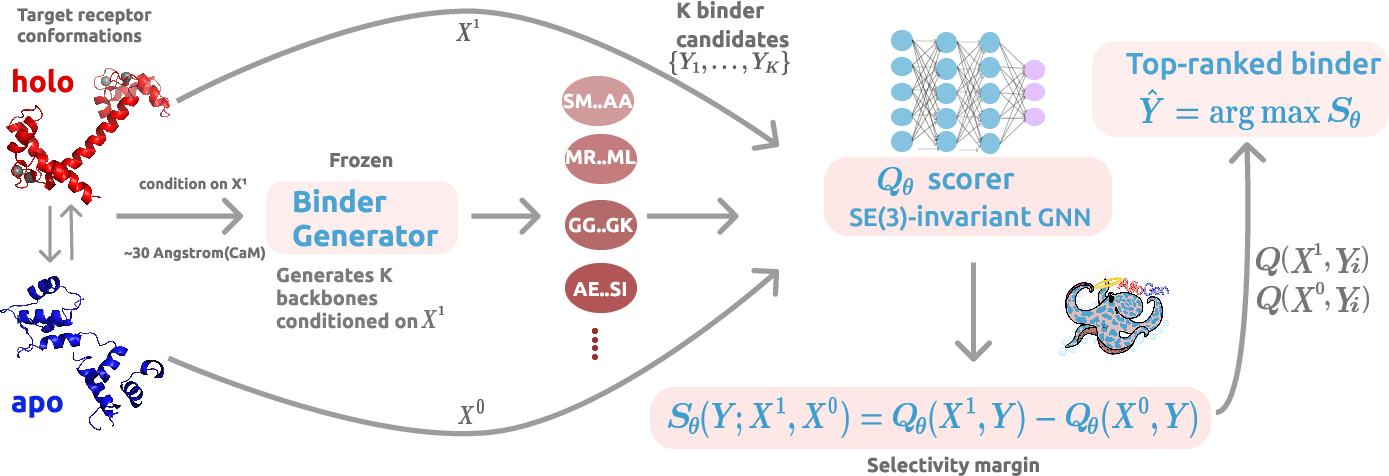
AlloGen: Conformation-Selective Binder Generation with Differential State Scoring
Abstract: Protein binder design has largely optimized for affinity alone, leaving conformational selectivity unaddressed: for allosteric targets such as kinases, nuclear receptors, and GPCRs, a binder that engages both active and inactive states provides no functional specificity regardless of how tightly it binds. We introduce AlloGen, a modular framework that decouples backbone generation from a learned state-selectivity scorer $Q_θ$, an SE(3)-invariant interface graph transformer trained via a two-phase curriculum that first learns interface geometry before imposing conformational discrimination. Because $Q_θ$ is fully differentiable and generator-agnostic, it integrates with any backbone generator as a passive reranker or an active gradient-based guide without retraining. Across a diverse benchmark of proteins spanning multiple families and conformational mechanisms, AlloGen consistently identifies binders that preferentially recognize desired structural states while rejecting alternative conformations. Experimental validation on calmodulin further demonstrates that these computational selectivity signals translate to physical molecules, yielding de novo peptides that bind the desired holo conformation while exhibiting no detectable binding to the apo state. Together, these results establish conformational selectivity as a learnable property and provide a general framework for state-selective protein binder design.

Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper is about teaching computers to design tiny proteins or peptides that stick to a target protein only when the target is in the “right” shape. Many proteins act like switches: they change shape to turn processes on or off. If a designed binder grabs both shapes, it can’t control anything. The authors created a system called AlloGen that helps design binders that prefer one shape (state) and avoid the other.
What questions were the researchers trying to answer?
They focused on three simple questions:
- Can we train a model to tell whether a binder prefers one shape of a protein over another?
- Can that model help different design programs make binders that are truly state-selective (pick the “on” shape and ignore the “off” one, or vice versa)?
- Do the model’s computer predictions match what happens in real lab experiments?
How did they do it?
Think of a protein like a machine that can be “open” or “closed.” A binder is like a custom glove that should fit only one version of that machine. AlloGen is a two-part setup:
- A generator: existing tools that propose possible binder shapes (like inventing many glove designs).
- A scorer called Qθ (say “Q-theta”): a learned “judge” that compares how well a binder fits the desired shape versus the undesired shape and gives a higher score when it prefers the right one.
Here’s how the key ideas work, in plain terms:
- Two states, one choice: Proteins often have at least two forms. The paper calls them apo (think “off”) and holo (think “on”). The goal is a binder that likes holo way more than apo.
- Learning to judge fit: First, Qθ is trained to understand “good interface quality” in general (like learning what a well-fitting glove looks like). This uses a standard quality score called DockQ, which measures how well two proteins fit together.
- Learning to tell states apart: Next, Qθ is fine-tuned with pairs of examples showing the same binder against two shapes of the same protein (on vs. off). It’s rewarded when it scores the correct shape higher. You can think of this like showing the judge two photos and asking, “Which fits better—and be consistent across many different proteins.”
- Ignore unimportant details: The judge learns to focus on geometry (the relative positions and directions of the parts) and not on where the protein sits in 3D space. That way, it scores the match the same even if you rotate or move the entire complex. This is what “SE(3)-invariant” means.
- Plug-and-play: Qθ can work with different binder-generating tools without retraining. It can:
- Rerank: pick the best designs out of a batch.
- Guide: gently push a designer tool toward better, more selective binders while it’s generating them.
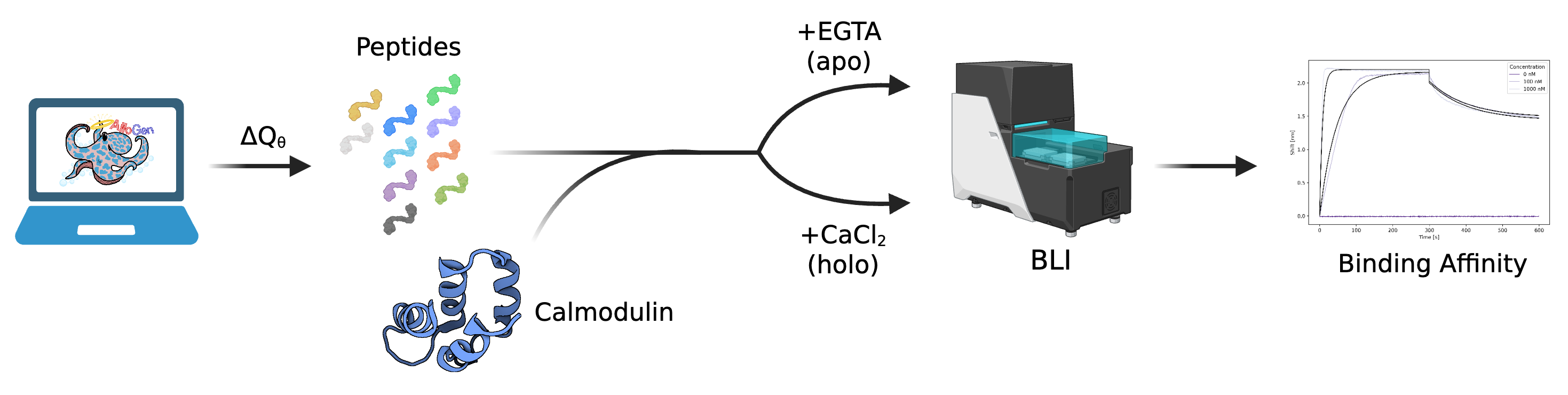
They tested AlloGen on many proteins with different types of shape changes and also did a real-world lab test with calmodulin (a protein that changes shape when it binds calcium).
What did they find, and why is it important?
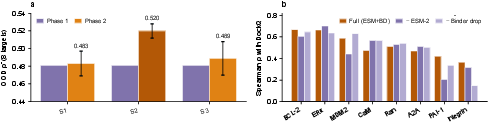
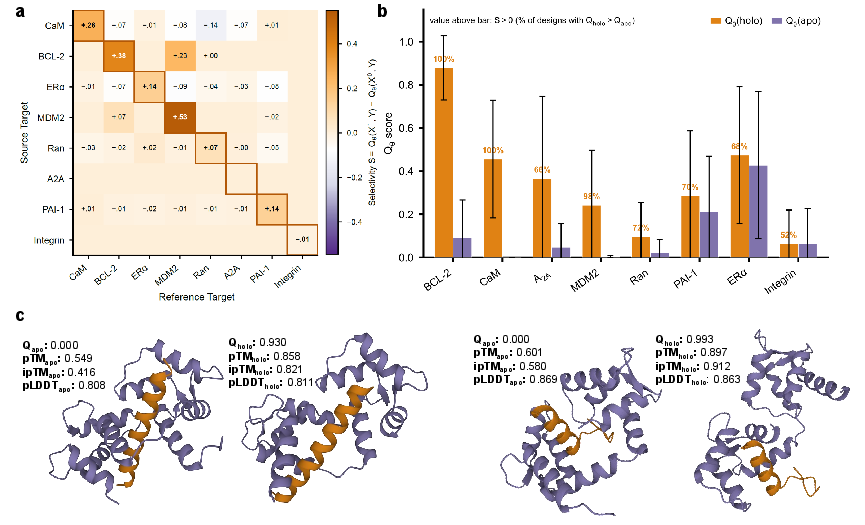
- The judge learned real selectivity: Qθ did a much better job than simple “contact” or “energy” scores at telling which binders truly prefer the desired shape over the undesired one. This means selectivity isn’t just “more sticks = better”—it’s about the right shape matching, and the model learned that.
- Works on new proteins: Qθ still worked on proteins it wasn’t trained on, which is important for designing binders for new targets.
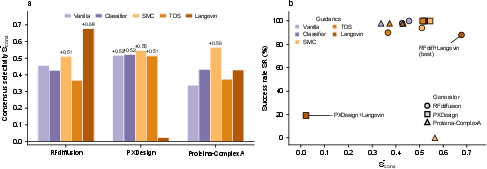
- Helps many design tools: Whether it just reranked results or actively guided the design process, Qθ pushed different generator programs to make more state-selective binders.
- Real lab success on calmodulin: Out of 10 designed peptides they tested, 5 bound strongly to calmodulin’s holo (calcium-bound, “on”) shape and showed no detectable binding to the apo (no-calcium, “off”) shape. A “negative control” design that scored poorly in the model didn’t bind at all. This shows the computer’s selectivity signal actually translates to real molecules.
Why this matters: In medicine and biology, we often want to control what a protein does, not just stick to it. Binding the wrong shape can cause side effects or do nothing useful. This method aims at the right kind of control.
What’s the big picture and future impact?
AlloGen shows that “conformational selectivity” (preferring one shape of a protein) is something a model can learn and use to design better molecules. This could:
- Lead to smarter drugs that stabilize only the helpful state of a protein, reducing side effects.
- Improve biosensors and synthetic biology tools that depend on proteins switching states.
- Plug into many existing design pipelines because the scorer is modular and doesn’t have to be retrained for each tool.
In the future, the authors suggest expanding to more than two states, building selectivity directly into sequence design, and targeting complex, medically important proteins where choosing the right shape is crucial.
Knowledge Gaps
Unresolved gaps, limitations, and open questions
Below is a single, consolidated list of concrete knowledge gaps and open questions that remain after this study.
- Limited experimental scope: validation was performed on a single target (calmodulin) with a small peptide panel; no tests on other protein families, larger protein binders, nanobodies, or antibodies.
- Binding mode confirmation is absent: no structural determination (co-crystal/cryo-EM/NMR) of designed binder–receptor complexes to verify the predicted state-specific interface and pose.
- Functional specificity not demonstrated: the study measures binding selectivity (Kd) but not functional outcomes (e.g., signaling modulation for GPCRs, kinase activity shifts, receptor transcriptional programs).
- No assessment in cellular or in vivo contexts: robustness of conformational selectivity under crowding, variable ionic strength, cofactors, PTMs, and cellular chaperones is untested.
- Negative/apo-state assay depth is shallow: “no detectable binding” lacks quantified upper bounds on Kd and does not explore alternative off-path states or partially active intermediates.
- Limited correlation analysis: no systematic correlation between predicted selectivity margin (Δq or Sθ) and measured affinities across the experimental panel; calibration curves are missing.
- Multi-state generalization is untested: although the margin extends to multiple undesired states, empirical evaluation considered a single undesired state per target; performance with >2 states or ensembles is unknown.
- Generator bottlenecks unresolved: targets like ERα and Integrin show weak design selectivity; specific generator-side modifications (architectures, conditioning, priors) needed to overcome these cases are not identified.
- Lack of co-optimization: Qθ is used post hoc; joint training or co-tuning of generators with Qθ (to reduce trajectory–gradient mismatch and target-specific failure modes) is not explored.
- Gradient reliability across generators: noise–gradient alignment was studied for RFdiffusion but not systematically for PXDesign, Proteina-ComplexA, or other priors; general guidance schedules remain ad hoc.
- Physical interpretability of Qθ is limited: outputs are bounded scores not calibrated to ΔΔG between states; mapping Qθ or Sθ to thermodynamic quantities remains unaddressed.
- Sequence–structure distribution shift: training uses ESM-2 and sequence features on the receptor; inference for de novo binders lacks sequence context; the impact of missing MSA/evolutionary signal on generalization is unclear.
- Sidechain-level physics are under-modeled: the scorer relies on backbone-local frames and sparse geometric features; explicit sidechain packing, hydrogen bonding, solvation, electrostatics, and cofactors (e.g., Ca2+) are not modeled.
- Membrane and environmental effects are omitted: performance on membrane proteins (e.g., A2A) is only computational; lipid environment, membrane electrostatics/thickness, and detergent/nanodisc conditions are not incorporated.
- Receptor-structure uncertainty is not handled: robustness of Qθ to modeled receptor states (e.g., AF/MD-derived apo/holo ensembles) versus crystal structures is not evaluated.
- Data coverage and bias: the 65-target dataset may be enriched for tractable systems with known binders; generalization to targets without known complexes or with rare conformational mechanisms remains uncertain.
- Decoy realism and leakage: synthetic decoys (GenDecoys) and cross-family negatives may not reflect real competing interfaces; systematic audits for family leakage or overfitting to decoy artifacts are not reported.
- Limited uncertainty quantification: per-complex confidence estimates for Qθ/Sθ are absent; ensembles are used for averaging but not to produce calibrated prediction intervals or abstention criteria.
- Score hacking/adversarial designs: safeguards against designs that exploit Qθ artifacts (without physical plausibility) are limited to basic geometric filters; more rigorous adversarial and stress tests are needed.
- Selectivity thresholding and normalization: Sθ thresholds are not calibrated across targets; per-target baseline normalization or adaptive thresholds to ensure cross-target comparability are missing.
- Hyperparameter sensitivity: key choices (8 Å graph cutoff, RBF bases, binder-side dropout rate, InfoNCE temperature/batch, feature ablations) lack systematic, target-agnostic sensitivity analyses.
- Intermediate-state learning: although Qθ tracks interpolated CaM states, training uses discrete apo/holo pairs; explicit supervision on intermediate ensembles or MSM-derived pathways is not leveraged.
- Scaling to larger binders: performance and guidance behavior with longer, multi-domain proteins, cyclic peptides, or constrained scaffolds are not characterized; designability vs. selectivity trade-offs are unknown.
- Sequence-level developability is unaddressed: aggregation, solubility, protease resistance, chemical stability, immunogenicity, and expression yields for designed sequences are not evaluated or optimized.
- Proteome-wide off-target risks: propensity of designed peptides to bind other proteins or unintended conformational states (especially for amphipathic helices) is not profiled computationally or experimentally.
- Cofactor and PTM dependence: state-selectivity conditioned on cofactors (ATP, GTP, lipids) or PTMs (phosphorylation) is not modeled; generalization to such multi-factor state control is open.
- Computational efficiency at scale: while some costs are reported, end-to-end throughput, cost–benefit trade-offs (reranking vs. guidance) and practical campaign design (thousands to millions of candidates) need clearer guidance.
- Target difficulty predictors are absent: no diagnostic features are proposed to predict when a new target will be “BCL-2-like” (easy) vs. “Integrin-like” (hard), hindering triage and resource allocation.
- Reliability of AF-based filters for peptides: use of ipTM/pLDDT to filter peptide complexes is known to be noisy; alternative orthogonal structure filters or physics-based validation are not benchmarked.
- Lack of public, standardized benchmarks: reproducible multi-state benchmarks with apo/holo pairs, standardized splits, and metrics for conformational selectivity are not released or formalized.
- Ethical and safety considerations: potential risks of generating high-affinity, state-selective binders for critical pathways (e.g., GPCRs/kinases) are not discussed, nor are safeguards for misuse mitigation.
Practical Applications
Immediate Applications
The following use cases can be deployed now by combining AlloGen’s state-selectivity scorer (Q_θ) with existing backbone generators and standard wet-lab validation. Each item lists sectors, suggested tools/workflows, and key assumptions/dependencies.
- Conformation-selective triage in protein/peptide binder pipelines
- Sectors: healthcare/biotech, pharma R&D, academia; software tools
- What: Plug
Q_θinto RFdiffusion, PXDesign, Proteina-ComplexA (or similar) to rerank or guide candidate backbones for allosteric targets (e.g., kinases, GPCRs, nuclear receptors). Select designs with large selectivity margins ΔQ = Q_θ(holo) − Q_θ(apo). - Tools/workflows:
- Reranking: best-of-K selection using
Q_θ(generator-agnostic, minimal engineering). - Guidance: Langevin refinement on completed backbones; or resampling strategies (SMC, TDS) during generation.
- Post-design sequence assignment with ProteinMPNN; structural checks with Boltz/AlphaFold; BSA/clash filtering; BLI/SPR for affinity.
- Assumptions/dependencies:
- Paired apo/holo (or undesired/desired) structures of the receptor are available and correctly aligned.
- Generator can sample viable interfaces for the target (generation bottleneck may dominate on some targets, e.g., subtle ERα H12 repositioning).
- In vitro conditions approximate the structural states used for design (e.g., ion/lipid/cofactor presence).
- Conformation-specific research probes for signaling proteins
- Sectors: academia, biotech tools
- What: Design short peptides that bind only the active or inactive state of research targets (e.g., Ras, CaM, MDM2, BCL-2) as biochemical probes to dissect pathway dynamics and allostery.
- Tools/workflows:
Q_θ-guided peptide design; in vitro validation (BLI/SPR); optional fluorescent labeling to create pull-down/FRET reagents. - Assumptions/dependencies: State structures reflect in vitro/in vivo conformations; probe must not perturb biology beyond desired stabilization.
- State-selective chaperones for structural biology
- Sectors: academia, structural biology (cryo-EM, X-ray crystallography)
- What: Generate binders that stabilize a specific conformational state (or intermediate) to improve particle homogeneity, trap rare states, or enhance crystallization.
- Tools/workflows:
Q_θ-guided design; complex re-folding to confirm state bias; binding/functional QC; incorporation as chaperones in EM or crystallography workflows. - Assumptions/dependencies: Access to state-resolved structures; binders do not artifactually remodel the targeted state.
- Conformation-specific biosensor components (prototype stage)
- Sectors: diagnostics (research-use), synthetic biology
- What: Create peptides that bind only the “on” state (e.g., Ca2+-bound CaM) to drive FRET or split-reporter assembly for live-cell or in vitro sensors.
- Tools/workflows:
Q_θdesign; reporter fusion and screening; cell-based validation. - Assumptions/dependencies: Coupling binding to a measurable signal requires linker engineering and calibration; cellular context may alter conformations.
- Software plug-ins and services for selectivity scoring
- Sectors: software, cloud platforms, CROs
- What: Offer
Q_θas a scoring/guidance module (API or plugin) integrated into existing CAD pipelines (Rosetta-based, RFdiffusion-based) to enable state-aware design. - Tools/workflows: Containerized reranker; batch scoring for best-of-K selection; SDKs for SMC/TDS/Langevin hooks; job orchestration in HPC/cloud.
- Assumptions/dependencies: Users supply PDBs for at least two states; compute and data governance in regulated environments.
- Educational and benchmarking kits for conformational selectivity
- Sectors: academia, training
- What: Turn the AlloGen pipeline and calmodulin example into a reproducible teaching module for courses in protein design and ML-for-biology.
- Tools/workflows: Provided Hugging Face model, example PDBs, notebooks to run reranking/guidance and basic wet-lab readouts.
- Assumptions/dependencies: Access to minimal wet-lab/biophysics equipment for validation is optional but beneficial.
Long-Term Applications
These use cases require further development, scaling, broader datasets (multi-state), or regulatory validation before routine deployment.
- Therapeutic development of state-selective biologics (peptides/proteins)
- Sectors: healthcare/biotech, pharma R&D
- What: Develop conformation-specific peptide/protein therapeutics (e.g., GPCR or kinase state-biased binders for functional selectivity, selective antagonism, or biased signaling).
- Tools/workflows: End-to-end AlloGen-guided pipelines; high-throughput sequence diversification; affinity maturation; manufacturability/PK and immunogenicity de-risking; in vivo efficacy.
- Assumptions/dependencies:
- Preclinical-to-clinical translation of peptides/proteins (stability, bioavailability) remains challenging.
- Regulatory acceptance of AI-designed modalities; IP strategy for designed sequences.
- Targeted degradation and state-selective proximity therapeutics
- Sectors: healthcare/biotech
- What: Use state-selective binders as warheads in protein degraders (e.g., ubiquitin ligase recruiters) or proximity-inducing biologics to degrade or modulate only the active/inactive state.
- Tools/workflows: Design of state-specific binder fused to E3 ligase/recruiters; cell assays for selectivity and efficacy; structural confirmation of state bias.
- Assumptions/dependencies: Intracellular delivery and stability of biologic degraders; robust demonstration of state-specific degradation in cells.
- Conformation-aware antibody/nanobody discovery
- Sectors: healthcare/biotech, diagnostics
- What: Extend the framework to scaffolded proteins (e.g., nanobodies, repeat proteins) for conformation-specific reagents and therapeutics.
- Tools/workflows: Adapt generators to antibody/nanobody scaffolds; train/select for state-selectivity using
Q_θ-like scorers; experimental panning biased by state. - Assumptions/dependencies: Generators for antibody-class scaffolds with compatible geometry control; wet-lab display systems tuned to conformational selection.
- Multi-state and pathway-aware design (beyond two states)
- Sectors: academia, pharma R&D, software
- What: Generalize from apo/holo to a landscape with multiple functional/intermediate states, optimizing binders for a target subset while rejecting others.
- Tools/workflows: Extend
S_θto multi-negative sets; curate multi-state structural ensembles (e.g., MD, cryo-EM classes); co-optimization with kinetic/thermodynamic objectives. - Assumptions/dependencies: Availability/quality of multi-state data; robust alignment across heterogeneous conformations; scalable training.
- High-throughput conformation-specific diagnostics and companion tests
- Sectors: diagnostics, precision medicine
- What: Develop immunoassay-like tests employing conformation-selective binders to detect disease-relevant active states (e.g., oncogenic Ras-GTP-like conformations, specific GPCR active states) as companion diagnostics.
- Tools/workflows: Stabilized binder production; assay engineering (ELISA/lateral flow); clinical validation.
- Assumptions/dependencies: Clinical assay sensitivity/specificity standards; validation cohorts; reproducibility across sample matrices.
- Industrial biocatalysis control via state-selective modulators
- Sectors: industrial biotechnology, bioprocessing
- What: Tune enzyme flux by stabilizing specific conformations (active/inactive) in metabolic pathways to optimize yields.
- Tools/workflows:
Q_θ-guided binders; fermentation-scale expression; process control integration. - Assumptions/dependencies: Effects in complex intracellular environments; large-scale stability and cost of peptide/protein additives.
- Agriculture and environmental biosensing
- Sectors: agtech, environmental monitoring
- What: Engineer conformation-specific biosensors for plant stress receptors or environmental analytes (via engineered host proteins whose activation states report on stimuli).
- Tools/workflows: Binder design, reporter coupling, field-deployable formats.
- Assumptions/dependencies: Robust performance outside controlled lab conditions; regulatory and biosafety considerations.
- Cross-modality extensions (sequence-first and small-molecule interfaces)
- Sectors: software, pharma R&D
- What: Integrate conformational selectivity directly into sequence generation models; explore analogous differential-state scoring to guide small-molecule docking/design.
- Tools/workflows: Co-training sequence/backbone models with multi-state objectives; hybrid docking with differential scoring.
- Assumptions/dependencies: Reliable mapping from backbone-level selectivity to sequence-level specificity; adaptation of geometry-based scorers to ligand-scale physics.
Common Assumptions and Dependencies Across Applications
- Data availability/quality: Paired state structures (apo/undesired vs. holo/desired) must be experimentally accurate and appropriately aligned; inclusion of relevant cofactors, ions, lipids, and PTMs is often necessary.
- Generator capability: Success depends on whether the generator can propose interfaces that exploit conformational distinctions; for some targets, generation (not scoring) is the bottleneck.
- Translation to function: Binding selectivity does not guarantee desired functional outcomes; stabilization of a state may not directly map to pathway modulation without further optimization.
- Experimental validation: In vitro confirmation (e.g., BLI/SPR) and in-cell/in vivo validation are required; conditions must recapitulate the designed conformational state.
- Safety, manufacturability, and regulatory pathways: Especially for therapeutics, immunogenicity, stability, delivery, and regulatory guidance for AI-designed biologics are critical.
- Compute and integration: Effective use requires access to GPU/CPU resources and integration into existing CAD pipelines; cloud/HPC deployment may be needed for large best-of-K or SMC/TDS campaigns.
These applications leverage AlloGen’s key innovations—a generator-agnostic, differentiable, SE(3)-invariant selectivity scorer and demonstrated translation to wet-lab binders—to move from affinity-centric to state-selective design across research and industry.
Glossary
- AlphaFold 3: A state-of-the-art protein structure prediction system used here to validate conformational preferences. "AlphaFold~3 confirmed holo preference on all 50 designs each for ALK and ER"
- AlphaFold2: An earlier deep-learning model for protein structure prediction used for co-folding and backpropagation-based design. "BindCraft co-folds target and binder by backpropagating through AlphaFold2"
- Allosteric: Referring to regulation or binding at a site distinct from the active site that modulates protein function. "for allosteric targets such as kinases, nuclear receptors, and GPCRs"
- Apo: The ligand-free or inactive conformational state of a protein. "apo (undesired) conformation "
- Apo-mismatch: A negative training example pairing a binder with the apo receptor conformation instead of the holo. "1 negative apo-mismatch complex $(X^0_{\mathrm{aligned}, Y^{\mathrm{native})$ with label $0.0$"
- Bio-layer interferometry (BLI): A label-free optical technique to measure biomolecular interactions and binding kinetics. "Binding affinity was characterized by bio-layer interferometry (BLI)"
- Boltz-2 ΔipTM: A validation signal from the Boltz-2 predictor measuring the difference in predicted inter-protein TMscore between states. "Boltz-2 ipTM correlated with selectivity on A2A () and CaM ()"
- Buried surface area: The solvent-inaccessible area at a protein–protein interface indicative of interface size. "whose buried surface area (computed with freesasa) exceeds $800$\,\AA"
- Cα (C-alpha): The alpha-carbon atom of an amino acid, commonly used to represent protein backbones. "target C RMSD levels spanning 1--8\,\AA"
- Calcium ion (Ca2+): A divalent cation whose binding can induce conformational changes (e.g., in calmodulin). "its 30\,\AA\ apo-to-holo rearrangement on Ca binding opens a hydrophobic peptide-binding cleft"
- Calmodulin (CaM): A calcium-binding protein used as an out-of-distribution validation target. "Experimental validation on calmodulin (CaM)"
- CD-HIT: A sequence clustering tool used to remove near-duplicate designs by identity thresholds. "then collapse near-duplicates by clustering the retained sequences at identity with CD-HIT"
- Classifier guidance: A diffusion guidance method that injects gradients from an external scorer to steer sampling. "classifier guidance rarely improving over vanilla sampling"
- Cross-chain edge density: A contact-based proxy measuring the density of inter-chain edges/residues at the interface. "cross-chain edge density all failed to track DockQ on average"
- DockQ: A composite metric in [0,1] for docking quality combining contact recovery and RMSD terms. "DockQ~\citep{basu2016dockq} is a composite scalar that measures protein--protein docking quality"
- Edge-biased graph transformer: A transformer architecture where edge features bias attention logits for structured graphs. " is implemented as a dense edge-biased graph transformer"
- EGTA: A calcium chelator used to prepare the apo form of calmodulin in experiments. "apo CaM (prepared with EGTA)"
- ESM-2 embeddings: Learned per-residue embeddings from a large protein LLM providing evolutionary context. "Among the input features, ESM-2 embeddings"
- freesasa: Software for computing solvent accessible surface areas, used here to estimate buried surface area. "buried surface area (computed with freesasa)"
- Gaussian RBF basis: A set of radial basis functions used to encode distances as smooth features. "the inter-residue distance via a Gaussian RBF basis"
- GenDecoys: Generator-produced synthetic decoy complexes used to provide hard negatives during training. "Augmenting the training set with GenDecoys, synthetic binders whose geometries span a broader region of interface space"
- GPCRs: G protein-coupled receptors, an important class of membrane proteins with multiple functional states. "kinases, nuclear receptors, and GPCRs"
- Holo: The ligand-bound or active conformational state of a protein. "holo (goal) conformation "
- Hotspot conditioning: Conditioning generators on interface-proximal residues (hotspots) from both states to focus design on relevant regions. "Dual-state hotspot conditioning."
- InfoNCE: A contrastive learning objective that separates positives from multiple negatives with a softmax normalization. "paired InfoNCE fine-tuning improved selectivity across all augmentation configurations"
- Interface RMSD: Root-mean-square deviation measured over interface residues to assess docking quality. "combining the fraction of native contacts, interface RMSD, and ligand RMSD"
- ipTM: Predicted inter-chain TMscore from structure predictors, used as an interface confidence metric. "per-state ipTM, pTM, and pLDDT"
- Kabsch-aligned: Alignment of two structures via the Kabsch algorithm to minimize RMSD. "where the apo receptor is Kabsch-aligned to the holo frame"
- KD (equilibrium dissociation constant): A measure of binding affinity derived from kinetic fits; lower values indicate tighter binding. "equilibrium dissociation constants () were determined from kinetic fits"
- Langevin refinement: A gradient-based post-generation refinement step applied to denoised backbones to increase the selectivity score. "Langevin refinement, by contrast, depended on the generator prior"
- Ligand RMSD: RMSD of the ligand (or binding partner) relative to a reference structure. "combining the fraction of native contacts, interface RMSD, and ligand RMSD"
- logit: The inverse-sigmoid transformation applied to probabilities, used here in defining the selectivity margin. "S$<em>\theta(Y;\, X<sup>1,</sup> \mathcal{N}) ={}& \mathrm{logit}\big(Q</em>\theta(X<sup>1,</sup> Y)\big) \ &- \log \sum_{X<sup>-</sup> \in \mathcal{N} ..."</li> <li><strong>Out-of-distribution (OOD)</strong>: Data or targets not seen during training, used to assess generalization. "eight out-of-distribution (OOD) test targets withheld during training"</li> <li><strong>pLDDT</strong>: Per-residue confidence score from AlphaFold-style predictors indicating local model reliability. "per-state ipTM, pTM, and pLDDT"</li> <li><strong>pTM</strong>: Predicted TMscore from AlphaFold-style predictors indicating overall structural confidence. "per-state ipTM, pTM, and pLDDT"</li> <li><strong>PRODIGY</strong>: A predictor of protein–protein binding affinities used as a baseline energy-based proxy. "PRODIGY~\citep{xue2016prodigy}"</li> <li><strong>ProteinMPNN</strong>: A neural network for protein sequence design conditioned on a fixed backbone. "sequence design is performed with ProteinMPNN"</li> <li><strong>PXDesign</strong>: A fast, modular protein binder generator used in the study’s benchmarks. "PXDesign delivers fast and modular binder design with strong experimental success rates"</li> <li><strong>REU (Rosetta Energy Units)</strong>: The internal energy unit used by Rosetta for scoring structures and interfaces. "assigned the most favorable interface energy to BCL-2 ($-9.1$ REU)"</li> <li><strong>RFdiffusion</strong>: A diffusion-based generative model for protein backbone design. "RFdiffusion establishes de novo design of functional protein binders at scale"</li> <li><strong>RMSD</strong>: Root-mean-square deviation between corresponding atoms used to assess structural similarity. "RMSD levels spanning 1--8\,\AA"</li> <li><strong>Rosetta FastRelax</strong>: A Rosetta protocol for constrained relaxation/repacking to generate near-native or decoy structures. "Rosetta FastRelax repacking on the holo receptor to produce near-native hard negatives"</li> <li><strong>Rosetta InterfaceAnalyzer</strong>: A Rosetta tool that evaluates protein–protein interface energies and metrics. "Rosetta InterfaceAnalyzer assigned the most favorable interface energy"</li> <li><strong>Ramachandran outliers</strong>: Residues whose backbone dihedral angles fall outside favored regions in the Ramachandran plot. "with zero Ramachandran outliers"</li> <li><strong>SE(3)</strong>: The group of 3D rigid motions (rotations and translations) relevant for geometric invariance. "for all rigid motions $g \in \mathrm{SE}(3)$"</li> <li><strong>SE(3)-invariant</strong>: Unchanged under any 3D rigid motion; here, a property of the scoring model and features. "an SE(3)-invariant interface graph transformer"</li> <li><strong>SO(3)</strong>: The group of 3D rotations used to express local backbone frames. "$R_i \in \mathrm{SO}(3)$"</li> <li><strong>Sequential Monte Carlo (SMC)</strong>: A particle-based resampling method used here to guide generation toward high-selectivity candidates. "Sequential Monte Carlo (SMC) iteratively resamples complete trajectories by $S_\theta$"</li> <li><strong>Spearman ρ</strong>: A nonparametric rank correlation coefficient used to evaluate scoring performance. "Spearman $\rho$"</li> <li><strong>Selectivity margin</strong>: A differentiable score difference favoring the desired state over undesired states. "The selectivity margin is defined as:"</li> <li><strong>Twisted diffusion sampling (TDS)</strong>: A diffusion reweighting approach that preserves the prior while favoring high-scoring samples. "Twisted diffusion sampling (TDS) reweights diffusion particles by $\exp(S_\theta)$"</li> <li><strong>Twin-Strep biosensor</strong>: A capture surface functionalized with Twin-Strep tag chemistry for immobilizing peptides in BLI. "immobilizing each peptide on a Twin-Strep biosensor"</li> <li><strong>ΔNLL (Delta NLL)</strong>: Difference in negative log-likelihood used to compare sequence preferences across states. "ProteinMPNN $\Delta\mathrm{NLL}$ favored holo on all eight targets for vanilla designs"
Collections
Sign up for free to add this paper to one or more collections.

