- The paper develops and validates a neural network-based ML interatomic potential for Ge-rich GST alloys, achieving energy RMSE <13.3 meV/atom and force RMSE <0.2 eV/Å.
- The study uses extensive atomistic simulations to replicate DFT-predicted structural, dynamical, and thermodynamic properties across amorphous, liquid, and crystalline phases.
- The MLIP enables large-scale simulations that capture crystallization and phase separation dynamics, offering critical insights for phase-change memory device modeling.
Atomistic Simulation of Ge-Rich GeSbTe Alloys via MLIP: Phase Separation and Crystallization
Machine Learning Interatomic Potential Construction and Validation
The study develops and rigorously validates a neural network-based machine learning interatomic potential (MLIP) for GeSbTe (GST) alloys, with a particular focus on Ge-rich compositions. The training database comprises ≈470,000 configurations, including amorphous, liquid, and crystalline phases, and interfaces generated using metadynamics. RMSEs for energies and forces across the training and test sets are consistently low (energy RMSE <13.3 meV/atom, force RMSE <0.2 eV/Å), indicating robust accuracy.
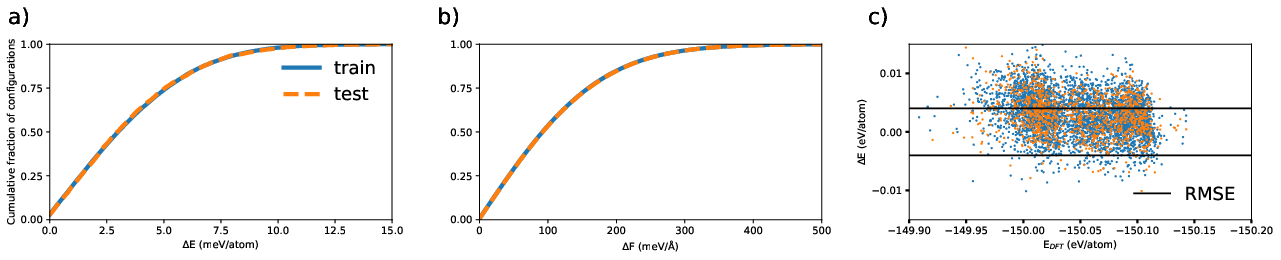
Figure 1: Cumulative fraction of NN potential absolute errors for GST523 training/test datasets, capturing energy and force deviations relative to DFT.
Validation proceeds through comparative molecular dynamics simulations using both MLIP and DFT across GST225, GST523, GeTe, Ge₂Te, GeSb, Ge₁₅Sb₈₅, Sb₂Te₃, Sb₂Te, and elemental systems (Ge, Sb, Te). Structural (radial/partial distribution functions), dynamical (diffusion coefficients), and thermodynamic properties (EOS) consistently exhibit strong quantitative agreement with DFT data. For example, in GST225 amorphous phase, the fraction of tetrahedrally coordinated Ge atoms is 22% (NN) vs 23% (DFT). EOS fits for crystalline phases yield equilibrium properties with minimal errors (<2% in bulk moduli).
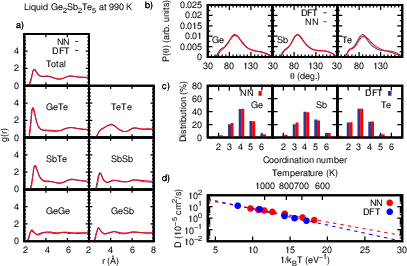
Figure 2: Structural and dynamical properties of liquid GST225 from NN-MD and DFT-MD simulations, validating MLIP accuracy across RDFs, angular distributions, and diffusion coefficients.
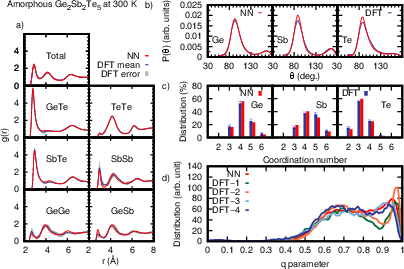
Figure 3: Structural properties of amorphous GST225 at 300 K, demonstrating close concordance between NN and DFT-derived RDFs, angular distributions, and tetrahedricity order parameters.
Large-Scale Simulation and Structural Characterization
The MLIP enables simulations of significantly larger supercells (up to 4096 atoms for GeTe) than feasible with DFT, facilitating robust statistical analysis and improved representation of low-abundance environments. Structural properties for both liquid and amorphous phases are characterized via radial and angular distribution functions, coordination number distributions, and local order parameters (such as q for Ge tetrahedricity).
Across tested compositions, MLIP predictions closely match DFT results, with occasional deviations (e.g., overestimation of tetrahedral Ge in Ge₁₅Sb₈₅: 81% NN vs 53% DFT). Typically, these discrepancies arise in low-concentration environments due to limitations in small DFT models. Transferability tests on compositions outside the training set (e.g., GST725, Ge₉.₆₂₅Sb₂Te₅, GST423, GST323) maintain strong structural fidelity.
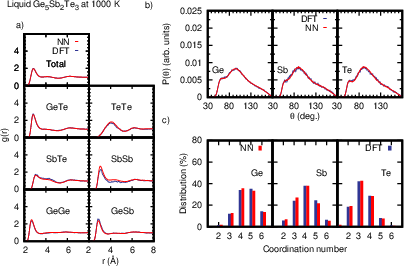
Figure 4: Structural properties of liquid GST523 at 1000 K from large-cell NN and small-cell DFT simulations, depicting RDFs, angular distributions, and coordination profiles.
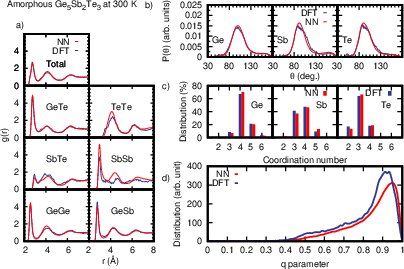
Figure 5: Structural properties of amorphous GST523 at 300 K from NN-MD and DFT-MD, including local tetrahedricity metrics for Ge coordination.
Thermodynamics and Phase Crystallization
The MLIP accurately reproduces equations of state for multiple crystal structures (cubic, trigonal, hexagonal), closely tracking DFT-computed energies, volumes, and bulk moduli. Crystallization kinetics are investigated via direct simulation of crystal-liquid interfaces, yielding crystal growth velocities and melting points consistent with experiment (e.g., GST225 melting temperature of 872 K NN vs 900 K experimental).
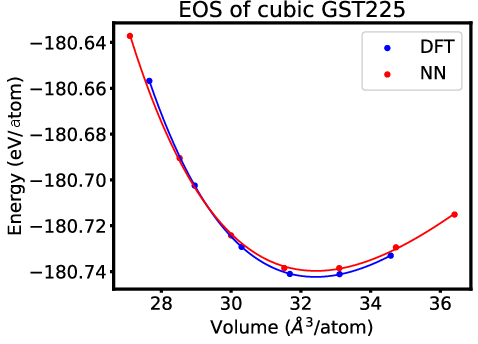
Figure 6: EOS of cubic GST225 via DFT and NN calculations, evidencing close energetic and volumetric matching.
Crystallization and Phase Separation in Ge-Rich GST
Crystallization and phase separation dynamics in Ge-rich GST725 were simulated using a large (8064-atom) cell. The process exhibits initial phase separation into GeTe-like and Ge-Sb-like amorphous regions, followed by crystal nucleation/growth within GeTe-like domains. Final products after ns-scale simulation are crystalline Ge₁₂SbTe₁₂ (slightly Sb-doped GeTe) and amorphous Ge₅₄Sb₄₆, diverging from expected thermodynamic equilibrium phases (Ge + GST225).
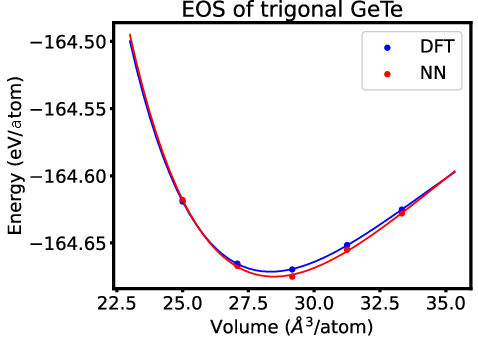
Figure 7: EOS of trigonal GeTe, verifying MLIP against DFT-PBE across crystal phases and yielding negligible energetic differences.
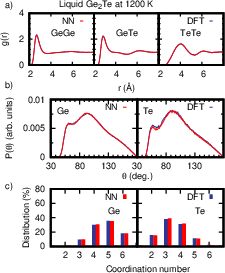
Figure 8: Structural properties of liquid Ge₂Te at 1200 K from NN-MD and DFT-MD, showing comprehensive agreement in distribution functions and coordination.
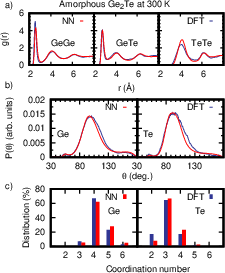
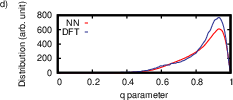
Figure 9: Structural properties of amorphous Ge₂Te at 300 K, illustrating MLIP’s accurate prediction of coordination environments and local tetrahedricity.
Implications and Future Directions
The implementation of MLIP for Ge-rich GST alloys demonstrates high accuracy, transferability, and computational scalability across a wide range of phases and compositions. The MLIP permits direct simulation of crystallization and phase separation processes at atomistic resolution and timescales inaccessible to DFT, thus enhancing mechanistic insight into microstructural evolution in phase-change materials.
Quantitative agreement with DFT supports MLIP’s utility for predictive modeling of GST alloys under varying thermodynamic conditions. Discrepancies observed in tetrahedricity fractions for highly Ge-abundant compositions suggest further refinement in database construction and model architecture for improved representation of rare environments.
Future work will likely explore incorporation of explicit van der Waals corrections, expansion to multi-component alloys, accelerated dynamics, and integration with experimental data to refine predictive performance for device-level phase-change applications.
Conclusion
The study presents a comprehensive MLIP framework for the simulation of Ge-rich GeSbTe alloys, validated against high-fidelity DFT data across structural, dynamical, and thermodynamic properties. The MLIP enables large-scale, atomistically resolved simulation of crystallization and phase separation, providing robust insight into microstructural transformations relevant for phase-change memory technologies and advancing the computational modeling landscape for GST alloys.