- The paper introduces an innovative integration of ITHC in AFQMC, enabling efficient diagonalization of the electron repulsion integrals via fictitious modes.
- It demonstrates reduced computational scaling and memory usage by transforming the two-body interaction into a diagonal form, optimizing GPU and matrix operations.
- Benchmark studies on H10 and benzene show chemical accuracy and controlled error extrapolation, confirming the method's potential for large-scale simulations.
Efficient AFQMC with Isometric Tensor Hypercontraction: Methodology and Applications
Introduction and Context
Auxiliary-Field Quantum Monte Carlo (AFQMC) stands as a state-of-the-art framework for simulating the electronic structure of molecular systems, offering a non-perturbative, systematically improvable treatment of electron correlation with lower polynomial scaling than traditional wavefunction-based methods. A primary bottleneck is the efficient treatment of the two-body electron repulsion integrals (ERI), often mitigated by Cholesky decomposition or approximate factorizations such as Tensor Hypercontraction (THC). The presented work innovates by integrating an Isometric Tensor Hypercontraction (ITHC) approach for representing the two-body operator, which enables efficient diagonalization via fictitious fermionic modes. This construction directly facilitates the use of a Hubbard-Stratonovich (HS) transformation within an extended Hilbert space, yielding a form suitable for both energy estimation and walker propagation with reduced theoretical and practical complexity.
The manuscript develops a rigorous extension of AFQMC wherein the two-electron operator is expressed as:
Vpqrs=α,β=1∑NauxupαuqαWαβurβusβ
with Naux≥N and u isometries connecting the physical orbital space to an enlarged auxiliary space. The introduction of fictitious modes, via the isometric extension, transforms the original two-body interaction into a diagonal "Hubbard-like" form:
W^=21α=β∑Wαβn^αn^β
where n^α operates in the extended space. This form is crucial for enabling an efficient and exact HS transformation as all number operators commute, and all steps (propagation, energy evaluation, force bias computation) become amenable to highly optimized matrix operations.
Algorithmic Implementation
The core computational step is the imaginary-time propagation, realized with a second-order Trotter-Suzuki decomposition combined with quantum Zeno dynamics to preserve projection onto the physical Hilbert space. Each propagation step involves:
A salient feature is the theoretical reduction in computational scaling: the dominant cost for propagation and evaluation of the local energy is reduced from O(NwNauxN2) and O(NwNauxNNe2) (in standard Cholesky-based implementations) to O(NwNauxNNe) and Naux≥N0, respectively. Memory usage is similarly reduced to Naux≥N1 due to the compressed storage of the Naux≥N2 matrix.
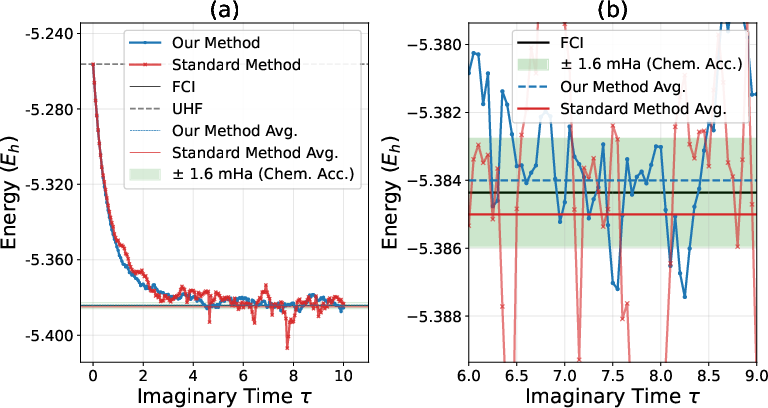
The AFQMC-ITHC method is benchmarked on the linear hydrogen chain (Naux≥N3). With STO-6G and UHF trial guesses, the computed ground-state energies closely match FCI results within chemical accuracy. Notably, the ITHC-based approach systematically overestimates the energy (compared to mild underestimation in standard AFQMC), and exhibits reduced stochastic noise, indicating improved sampling efficiency in the extended Hilbert space.
For larger systems, particularly the benzene molecule in the cc-pVDZ basis, the method robustly reproduces many-body correlation energies. By systematically varying the time step Naux≥N4, the authors demonstrate controlled extrapolation of the Zeno error, achieving a final correlation energy for benzene of Naux≥N5 mNaux≥N6—within 0.2% of the canonical MBE-FCI benchmark and outperforming high-level CCSDT.
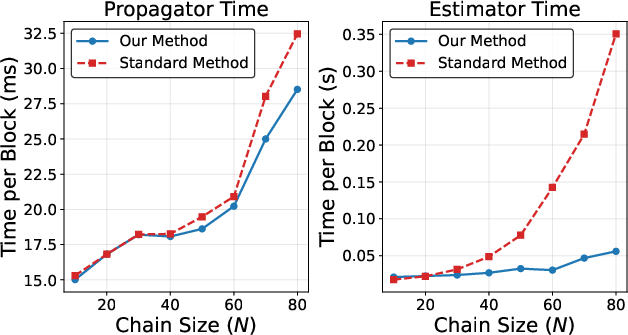
Figure 2: GPU runtime benchmarks for AFQMC propagation and estimator evaluation, highlighting the superior scaling and raw performance of the ITHC-based implementation versus standard AFQMC for increasing hydrogen chain length.
Performance comparisons reveal that, for moderate and large system sizes, the method yields significant reductions in both wallclock propagation and estimator time when executed on GPU hardware, with particularly marked improvements in the estimator phase for Naux≥N7.
Zeno Dynamics and Error Scaling
A central theoretical aspect is the analysis of numerical errors. The Trotter error remains subleading (Naux≥N8), while the main contributor to systematic bias is the Zeno error (Naux≥N9), which arises from alternating evolution and projection within the extended Hilbert space. The error exhibits clear linear dependence on u0, facilitating straightforward extrapolation. In practice, the magnitude of the Zeno term is correlated with the operator norm of u1, and is further influenced by the choice and accuracy of the ITHC representation.
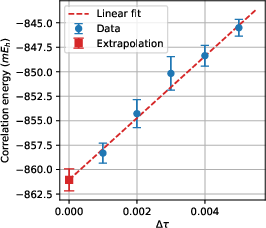
Figure 3: Linear extrapolation of the correlation energy for benzene as a function of u2, demonstrating control and removal of the Zeno error, and yielding a value in quantitative agreement with MBE-FCI and DMRG.
Implications and Outlook
This work provides an efficient framework for integrating advanced tensor factorizations, specifically ITHC, into AFQMC, facilitating high-precision ground-state electronic structure calculations for strongly correlated molecular systems. The reduction in scaling and memory footprint extends the frontier for tractable QMC simulations of larger molecules on modern GPU architectures. The approach maintains compatibility with existing AFQMC enhancements (multi-determinant trial states, improved importance sampling, etc.), pointing to direct synergy with recent algorithmic advances in both quantum chemistry and condensed matter.
However, single-determinant trial wavefunctions remain a limiting factor for systems with strong static correlation; future developments must focus on scalable generation and handling of multi-determinant trial states within the present framework. Additionally, adapting higher-order error cancellation schemes for Zeno dynamics, as recently explored in quantum simulation, holds promise for further systematic error reduction without sacrificing favorable scaling.
Conclusion
This paper presents a principled extension of AFQMC leveraging ITHC, resulting in polynomial reductions in computational and memory complexity while retaining or exceeding the accuracy of established wavefunction methods for correlated electronic systems. The approach is validated on both model and realistic systems, and the lower scaling paves the way for systematic studies of larger molecules. Theoretical control over error sources (Zeno, ITHC, Trotter) is demonstrated, and practical performance gains are substantiated numerically. The method offers a robust platform for future investigation of advanced trial states, error mitigation, and broader application of AFQMC in computational chemistry and materials science.