- The paper demonstrates that in-situ XPCS reveals coexisting fast domain wall motion and slow aging-dependent de-pinning during the LSCO phase transformation.
- The analysis quantifies distinct kinetic regimes with a fast relaxation (~10^3 s) and a slow, aging process that accelerates due to diffusion limitations.
- The study highlights the importance of defect-induced nucleation and tailored reduction protocols for optimizing LSCO-based phase-change device performance.
Introduction
This work investigates the dynamic spatial and temporal heterogeneity inherent to the perovskite-to-brownmillerite (P-to-B) topotactic phase transformation in La0.7Sr0.3CoO3 (LSCO) thin films under reducing conditions, utilizing in-situ Bragg X-ray photon correlation spectroscopy (XPCS) and X-ray diffraction (XRD). Exploiting the high sensitivity and temporal resolution of XPCS, the authors directly probe nanoscale dynamics during phase evolution, revealing complex aging behavior and diffusive limitations that cannot be resolved by classical diffraction methods alone. The study demonstrates the coexistence of multiple kinetic regimes during the P-to-B transformation and links these to underlying mechanisms such as domain wall motion, domain de-pinning, and oxygen ion diffusion.
The phase diagram of cobaltite perovskites such as LSCO is rich, with functionality tunable by epitaxial strain, oxygen stoichiometry, and external fields. The ability to reversibly switch between perovskite and brownmillerite phases, characterized by substantial changes in structural, electronic, and magnetic properties, is critical for future device applications in neuromorphic computing, spintronics, and energy conversion.
Experimental Methodology and Characterization
A 20 nm epitaxial LSCO film on LSAT substrate was studied using coherent synchrotron X-rays. Thermal annealing was performed under vacuum (P<1×10−3 mbar), with temperatures from room temperature to 550∘C. At each temperature increment, XRD and extended XPCS measurements were performed on both perovskite and brownmillerite Bragg peaks.
The coherent X-ray setup enabled the observation of speckle patterns in the half-order BM(006) Bragg peak, signaling the presence and evolution of brownmillerite domains within the beam footprint. XRD provided complementary ensemble-averaged structural information, while XPCS delivered direct access to slow dynamics and correlated nanoscale motion.
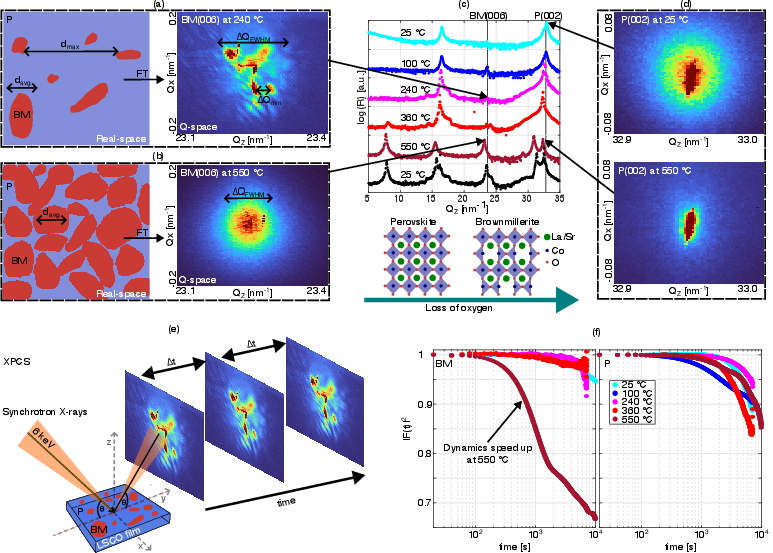
Figure 1: Coherent X-ray setup and Bragg peaks illustrate the domain structure evolution and the detection of dilute and dense brownmillerite phases as a function of temperature.
Static Phase and Domain Structure Determination
XRD confirmed the expected phase transformation from perovskite to brownmillerite at elevated temperature in reducing conditions, with the brownmillerite half-order peak clearly resolved only in synchrotron measurements (Figure 1). At temperatures below the phase transition, the LSCO films contain trace amounts (approximately 0.2%) of brownmillerite domains, as estimated from speckle pattern analysis and Bragg peak characteristics. The average domain size prior to transition is approximately 60 ± 5 nm. Above the transition, the domain structure becomes more homogeneous, and the characteristic BM domain size remains on the order of 55 ± 5 nm.
The initial presence of brownmillerite within the perovskite matrix suggests that nucleation at pre-existing domains or defects plays a substantial role in the transformation kinetics. This is consistent with prior nano-diffraction findings and highlights the importance of defect engineering for deterministic control of transformation pathways.
Spatiotemporal Dynamic Heterogeneity Uncovered by XPCS
XPCS revealed marked temporal heterogeneity in the phase transformation, with two separable relaxation timescales associated with different aspects of the brownmillerite domain evolution. Two-time correlation analysis at 550∘C demonstrates a fast relaxation process (τf∼103 s) likely reflecting domain wall motion and a slower relaxation timescale (τs) exhibiting pronounced aging dependence, accelerating by almost an order of magnitude over approximately 9000 s, well described by a power law decay with exponent −2.2±0.5.
(Figure 2)
Figure 2: (a) Two-time correlation function G(t1,t2) at 550∘C and (b) corresponding age-dependent intermediate scattering functions showing emergence of a slow dynamical process with system aging.
Fits to stretched exponential models yield a stretching parameter β=1.6±0.3 consistent with glassy/jammed dynamics, and an initial contrast of 18%, indicating that a minority of the domain area is dynamically active at early stages. With increasing aging time, the contrast increases, signifying a growing fraction of the dynamic phase.
(Figure 3)
Figure 3: (a) Evolution of τs and τf with aging; τs accelerates strongly, while τf remains relatively stable. (b) Contrast increase indicates an expanding dynamically active volume fraction during aging.
Domain wall velocities were estimated at vd=6±0.5×10−4 nm/s (2 ± 0.2 nm/h), and the slow domain de-pinning rate further reinforces the fundamentally diffusion-limited transformation kinetics.
Mechanistic Interpretation and Broader Relevance
The persistence of slow dynamical aging phenomena, even after the apparent completion of the bulk transformation, suggests significant kinetic bottlenecks imposed by domain de-pinning and long-range glassy interactions. The coexistence of time-independent fast and aging slow processes points to distinct mechanisms: rapid domain wall advancement (likely limited by local energy barriers and oxygen mobility) and the sluggish collective motion of domains as they detach from pinning defects. The latter accelerates as the system ages and structural relaxation occurs.
Comparison with prior electrochemical gating and switching experiments in LSCO devices indicates that XPCS-sensitive timescales match those observed in functional scenarios, especially in the context of device ON/OFF switching associated with phase-pure brownmillerite formation. Thus, the nanoscale dynamics resolved here offer direct insight into the ultimate speed limitations for reconfigurable oxide electronics.
For phase-change and correlated oxide device applications, these findings underscore that achieving instantaneous switching is precluded by the inherent glass-like aging dynamics in the solid-state transformation process. Dynamic heterogeneity and slow domain evolution must be considered in device design, both as possible performance bottlenecks and as sources of stochasticity.
Theoretical and Practical Implications
The demonstrated sensitivity of XPCS to both nucleation/growth and collective de-pinning mechanisms solidifies its position as a critical technique for interrogating the slow dynamics in topotactic transitions, complementing structural and transport measurements. The power law acceleration of slow timescales echoes universal features observed near glass transitions and in other percolative systems, suggesting broader applicability for describing complex solid-state transformations beyond LSCO.
Optimizing device architectures will thus require strategies for mitigating dynamic heterogeneity—possibly by engineering defect landscapes or tuning reduction protocols to homogenize nucleation and reduce pinning. The potential for direct measurement of these effects under device operation or in operando switching environments is suggested.
On a methodological level, integration of XPCS with ultrafast X-ray sources could extend access to regimes from seconds to picoseconds, providing a comprehensive view of activation dynamics across all relevant timescales.
Conclusion
This study elucidates the nanoscale spatiotemporal dynamics of the P-to-B phase transformation in LSCO thin films, distinguishing between fast domain wall motion and much slower, aging-dependent domain de-pinning and rearrangement—both ultimately limited by oxygen ion diffusion. The presence of preformed nuclei and significant kinetic bottlenecks highlights the need for advanced nanoscale probing strategies in assessing and engineering functionality in correlated oxide systems. The results have immediate implications for the design and performance limits of LSCO-based phase-change and neuromorphic devices and demonstrate the unmatched capability of XPCS for characterizing dynamic heterogeneity in complex oxides.

