- The paper demonstrates that a Transformer-based energy model can effectively predict atomic-resolution protein conformations using crystallized protein data.
- The model utilizes Cartesian coordinate embeddings and categorical features processed through Transformer blocks to capture nonlinear atomic interactions.
- Experimental results show competitive rotamer recovery rates compared to the Rosetta energy function, with distinct performance on small and polar amino acids.
Introduction
The paper "Energy-based models for atomic-resolution protein conformations" (2004.13167) presents an innovative approach to predicting protein conformations at an atomic scale using energy-based models (EBMs). Unlike traditional methods incorporating physical knowledge and complex features accumulated over decades, the proposed model is trained directly on crystallized protein data, demonstrating comparable performance to existing state-of-the-art methods such as the Rosetta energy function. The approach leverages the Transformer architecture to evaluate molecular configurations, particularly in the rotamer recovery task, where the conformation of side chains within protein structures is predicted.
Methodology
Model Architecture
The core of the methodology is the Transformer-based EBM, termed Atom Transformer. The model accepts a subset of atoms as input, embedding their Cartesian coordinates and categorical features. Sequences of embeddings pass through Transformer blocks, concluding in a two-layer multilayer perceptron (MLP) outputting a scalar energy value, fθ(A).
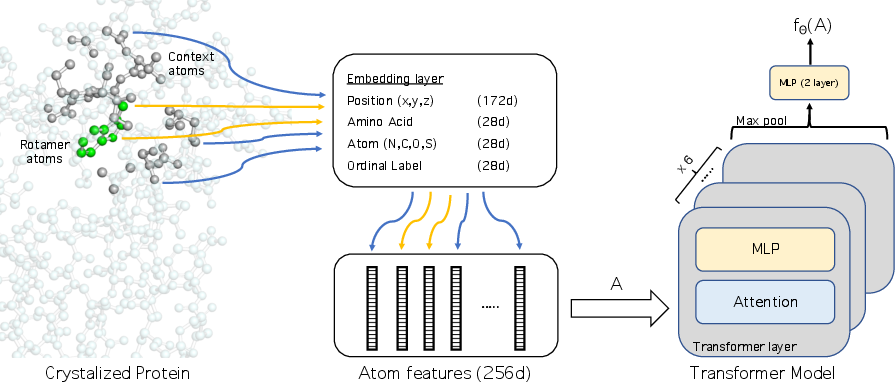
Figure 1: Overview of the model. The model processes a set of atoms to output a scalar energy value.
To define the atom subsets A, the model selects k nearest atoms to the residue's beta carbon. Three categorical features—identity, ordinal position, and amino acid type—alongside normalized spatial coordinates, form the atom input representation. The Transformer architecture enables modeling of nonlinear interactions among single and pairwise atom dependencies.
Training and Optimization
EBMs, characterized by their scalar parametric energy functions Eθ(x), apply maximum likelihood methods to minimize KL divergence between empirical data distributions and model distributions. Here, importance sampling draws from a rotamer library, approximating distributions via mixtures of Gaussians interpolated across backbone ϕ and ψ angles.
Experimental Evaluation
Dataset and Baselines
The dataset comprises high-resolution PDB structures curated under stringent conditions, including low sequence identity and resolution thresholds. Comparisons are made against baselines (fully-connected, set2set, graph neural networks) with comparable parameter counts, and Rosetta energy functions under discrete and continuous sampling schemes.
The Atom Transformer demonstrates robust performance, achieving accuracy near Rosetta in rotamer recovery rates. Specifically, the model exhibits better results on small and polar amino acids, while Rosetta excels on larger residues like phenylalanine and leucine.
Table comparisons reveal the Atom Transformer's competitive edge in rotamer recovery under both discrete and continuous methods.
Further Insights
Distinct behavior between core and surface residues is captured by the energy function, manifesting through differential sensitivity to perturbations in χ1 torsion angles.

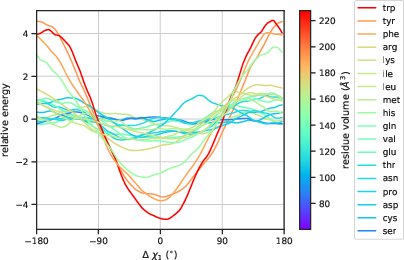
Figure 2: Core residues show heightened sensitivity to perturbations in χ1 torsion angles.
Additionally, visualizations expose clustering tendencies in representations of core versus surface residues and identify symmetry in amino acid responses to angle variations.
Implications and Future Work
This paper sets a foundation for future advancements in protein design by proposing a model that automatically learns features from data. The potential for generalization to more complex scenarios like side chain combinatorial optimization and inverse folding indicates a promising trajectory for designing innovative proteins. While traditional physical principles are integral for de novo design, the integration of data-driven neural models can accelerate novel protein discoveries, expanding beyond evolutionary landscapes.
Conclusion
The energy-based model detailed in the study offers a significant contribution to understanding protein conformational dynamics at an atomic level. By directly learning from crystallized datasets, this approach fosters exploration in uncharted territories of protein design, suggesting that generative models informed by empirical insights could transform future protein engineering endeavors.