SimpleFold: Folding Proteins is Simpler than You Think
Abstract: Protein folding models have achieved groundbreaking results typically via a combination of integrating domain knowledge into the architectural blocks and training pipelines. Nonetheless, given the success of generative models across different but related problems, it is natural to question whether these architectural designs are a necessary condition to build performant models. In this paper, we introduce SimpleFold, the first flow-matching based protein folding model that solely uses general purpose transformer blocks. Protein folding models typically employ computationally expensive modules involving triangular updates, explicit pair representations or multiple training objectives curated for this specific domain. Instead, SimpleFold employs standard transformer blocks with adaptive layers and is trained via a generative flow-matching objective with an additional structural term. We scale SimpleFold to 3B parameters and train it on approximately 9M distilled protein structures together with experimental PDB data. On standard folding benchmarks, SimpleFold-3B achieves competitive performance compared to state-of-the-art baselines, in addition SimpleFold demonstrates strong performance in ensemble prediction which is typically difficult for models trained via deterministic reconstruction objectives. Due to its general-purpose architecture, SimpleFold shows efficiency in deployment and inference on consumer-level hardware. SimpleFold challenges the reliance on complex domain-specific architectures designs in protein folding, opening up an alternative design space for future progress.

Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
Overview
This paper introduces SimpleFold, a new way to predict how proteins fold into their 3D shapes using a simpler kind of AI model than what most famous systems use. Instead of relying on many special, complex parts tailored to biology, SimpleFold uses general-purpose “transformer” blocks (the same kind of AI used in language and image models) and a generative training method. The big idea: protein folding can be done well without heavy, domain-specific tricks, making it faster, easier, and still accurate.
What is the paper trying to figure out?
The researchers asked:
- Do we really need complex, biology-specific model parts (like multiple sequence alignments and special geometry modules) to predict protein shapes?
- Can a simpler, general-purpose model generate accurate protein structures from just the amino acid sequence?
- Can such a model produce not just one shape, but an ensemble of possible shapes (because proteins can wiggle and take slightly different forms in nature)?
- Can this model be efficient enough to run on consumer-level hardware and still perform competitively?
How did they approach the problem?
Key ideas in simple terms
- Proteins are chains of amino acids that fold into detailed 3D shapes. Predicting that shape from the chain is called “protein folding.”
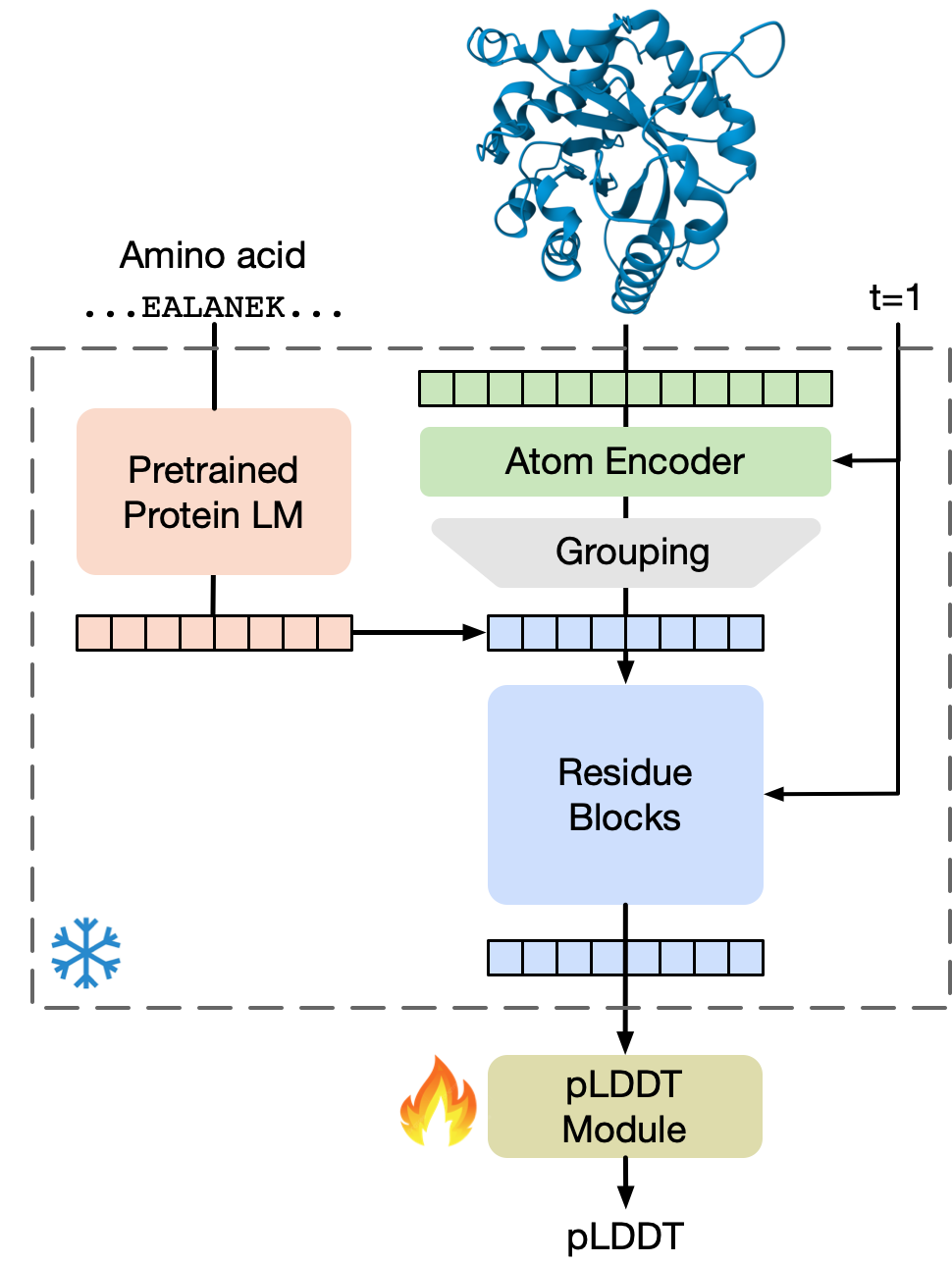
- SimpleFold treats folding like “text-to-image” generation: the amino acid sequence is the “prompt,” and the output is the 3D positions of all atoms in the protein.
- The model uses transformer blocks, a general AI building tool that’s good at spotting patterns and relationships.
- Instead of copying special tricks from older models (like comparing many similar sequences called MSA, or using custom geometry modules), SimpleFold keeps the architecture simple and lets the model learn directly from data.
What is “flow matching” (in everyday language)?
Flow matching is a way to teach an AI to turn random noise into a meaningful result step-by-step. Imagine starting with a cloud of points (noise) and gradually nudging them into the correct 3D shape of the protein. The model learns a “velocity field,” which is like instructions telling each atom how to move at each timestep to go from noise to the final structure.
- Time t goes from 0 (very noisy) to 1 (clean structure).
- The model learns to predict the direction and speed each atom should move at each step.
- This generative process naturally captures uncertainty and can produce multiple plausible shapes (ensembles), not just one.
What data and features did they use?
- Training data included millions of protein structures:
- Experimental structures from the PDB (Protein Data Bank).
- High-confidence “distilled” structures from large public databases (AFDB, ESM Metagenomic Atlas, AFESM).
- They trained models of different sizes (from 100 million to 3 billion parameters).
- They used a protein LLM (ESM2) to turn amino acid sequences into useful numeric features, similar to how LLMs turn words into embeddings.
How does the model know if it’s doing well?
They used a local distance score called LDDT:
- LDDT checks how accurate local atomic distances are (think: “Are neighboring atoms the right distance apart?”).
- The model also predicts a confidence score called pLDDT for each residue (section of the protein chain), which says how reliable that part of the prediction is.
How does the model generate a structure?
- Start with random positions for all atoms (noise).
- Integrate the learned velocity field from t=0 to t=1 to “denoise” the positions into a realistic protein structure.
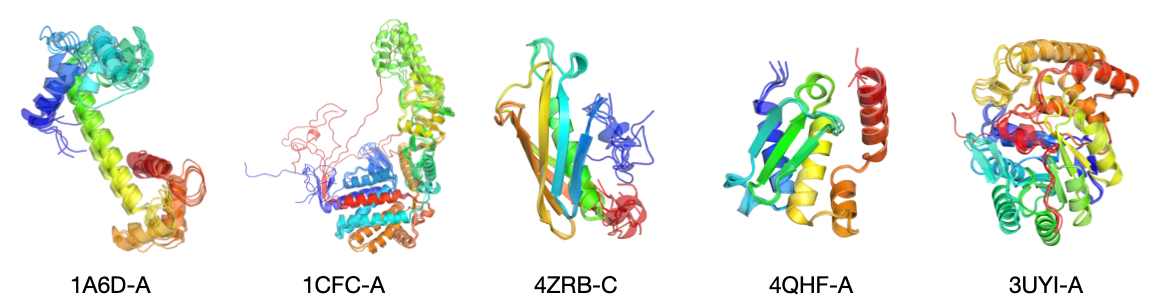
- This can be done repeatedly to create an ensemble (multiple conformations), which is useful because real proteins can flex and move.
What did they find?
Here are the most important results:
- Competitive accuracy without complex parts:
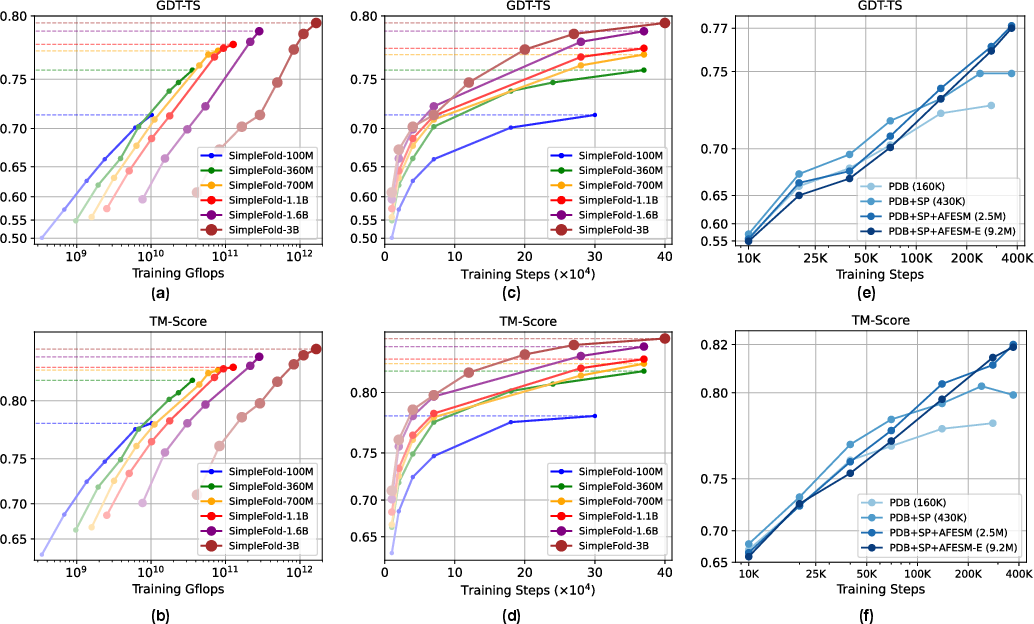
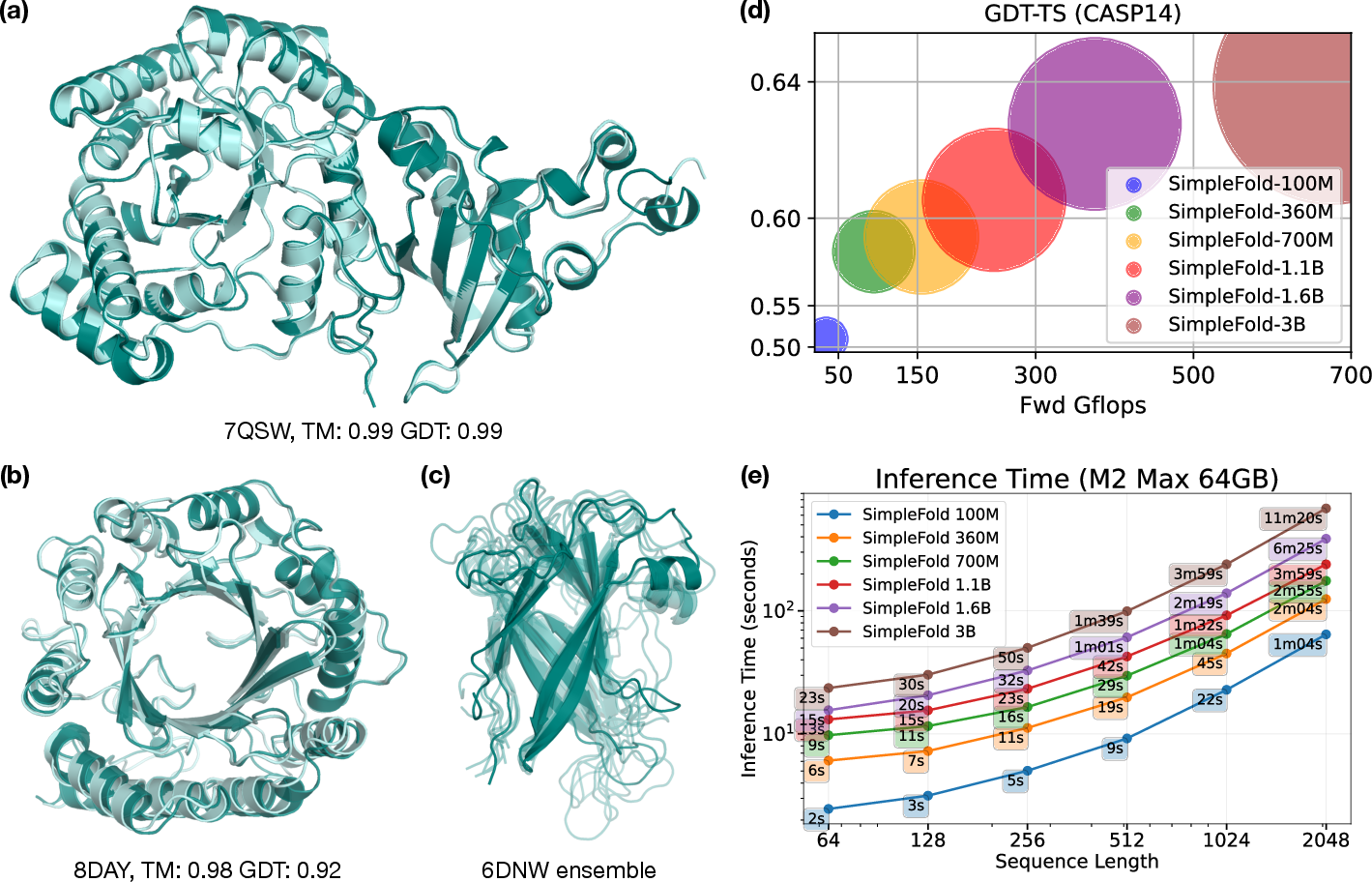
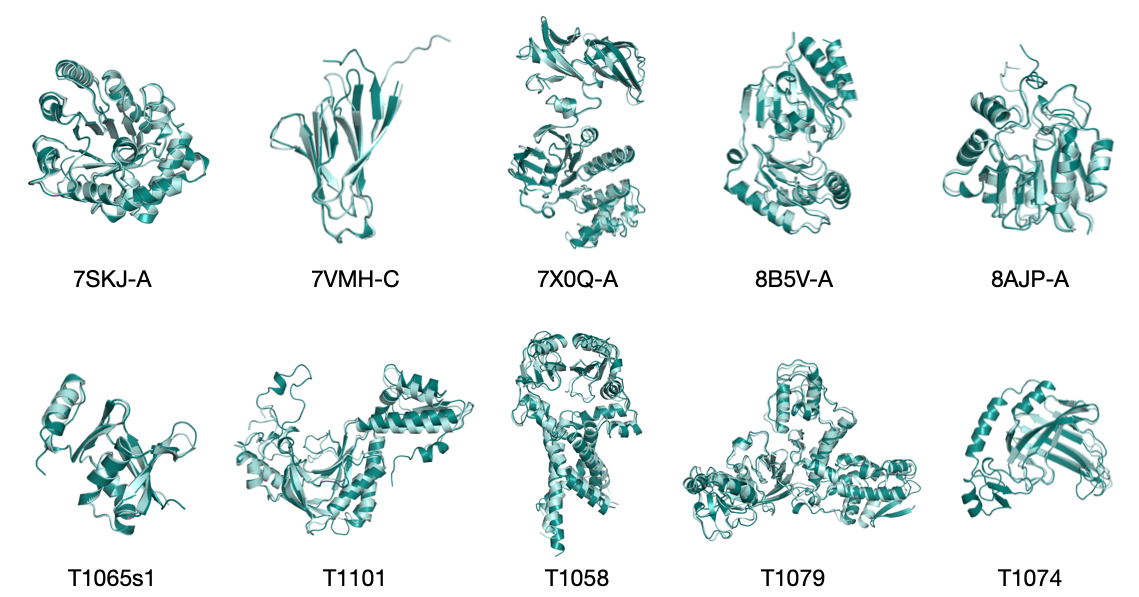
- On standard benchmarks (CAMEO22 and CASP14), SimpleFold performs competitively with well-known models, often reaching over 90–95% of the best scores, even without using MSAs or triangle updates.
- On CASP14 (a tougher test), SimpleFold’s largest model beats ESMFold (a strong baseline without MSA) on key metrics like TM-score and GDT-TS.
- Strong at ensemble prediction:
- SimpleFold naturally generates multiple valid shapes (ensembles), which is hard for models trained to produce just one solution.
- On molecular dynamics (MD) ensemble benchmarks, SimpleFold performs well and improves further when fine-tuned on MD data.
- Scales with size and data:
- Larger models trained on more data generally performed better, especially on challenging tasks.
- Efficient and practical:
- Because it uses general-purpose transformers and avoids heavy, custom modules, SimpleFold is simpler to deploy and can run on consumer hardware (like an M2 Max MacBook Pro).
- Confidence estimation:
- The pLDDT module gives per-residue confidence scores that correlate well with actual accuracy, helping users judge reliability of the predicted structures.
Why is this important?
- Simplicity matters: The paper shows you can get strong protein folding performance with a simpler, more general AI design. This challenges the idea that you must rely on complex, domain-specific modules to succeed.
- Generative understanding: By modeling folding as a generative process, SimpleFold captures real-world uncertainty and can output multiple plausible shapes—which is closer to how proteins behave in nature.
- Wider access: Simpler models that run on everyday hardware make advanced protein prediction more accessible to more researchers, students, and labs.
- Faster progress: A general-purpose approach can benefit from advances in transformer architectures and generative modeling without re-engineering complex biological modules each time.
- Impact on drug discovery and biology: Better, faster folding and ensemble prediction can help find proteins’ flexible, functional forms, identify new drug targets, and speed up design and discovery.
Final takeaway
SimpleFold suggests that folding proteins may be “simpler than you think.” By using a straightforward transformer-based generative model and lots of training data, it achieves competitive accuracy, produces realistic ensembles, and runs efficiently—without relying on heavy, specialized architecture. This opens up a new design space for protein folding models and could make cutting-edge biology tools more scalable, flexible, and accessible.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a single, consolidated list of what remains missing, uncertain, or unexplored in the paper. Each point is phrased to be directly actionable for future research.
- Generalization to multimers and complexes is untested: the model is evaluated only on single-chain proteins; performance on protein-protein, protein–nucleic acid, and protein–ligand complexes (including interface geometry and docking) remains unknown.
- Physical plausibility of all-atom outputs is not audited: no metrics are reported for bond-length/angle/dihedral violations, chirality, steric clashes, rotamer plausibility, Ramachandran outliers, or energy-based assessments; adding geometry checks and energy minimization evaluations is needed.
- Equivariance is not guaranteed: the non-equivariant transformer relies on SO(3) augmentation; the extent to which learned symmetry is complete, robust, and stable across lengths/topologies remains unquantified (e.g., controlled rotation tests and invariance stress tests).
- Long-range atomic interactions in atom-level modules may be under-modeled: atom encoder/decoder use local attention masks; the impact on long-range side-chain packing and distant allosteric interactions is not assessed.
- Sequence length constraints and truncation are limiting: pre-training uses max length 256, finetuning 512 (cropping longer sequences); systematic evaluation on very long proteins (>1,000 residues), multi-domain assemblies, and interdomain orientation accuracy is missing.
- Side-chain accuracy is not directly evaluated: while LDDT is reported, rotamer correctness (e.g., χ-angle accuracy), side-chain clash rates, and rotamer library consistency are not analyzed.
- Reliance on distilled structures may import biases from AF2/ESM pipelines: the model is trained on millions of predicted structures (AFDB/ESM/AFESM-E), which encode MSA-derived biases; the consequences for learning spurious patterns, generalization, and calibration are not studied.
- Dataset contamination and overlap controls are unclear: although a PDB cutoff is mentioned, thorough de-duplication across AFESM-E training data versus CASP/CAMEO test sets is not documented; rigorous leakage analyses are needed.
- No stratified evaluation on orphan proteins: the paper cites PLM benefits for orphans but provides no stratified results separating homolog-rich vs orphan targets to validate the claim.
- Benchmark scope is limited: comparisons omit recent generative folding models for complexes (e.g., AF3 and reproductions) and structure refinement baselines; broader baselines and standardized evaluation protocols would strengthen conclusions.
- Thermodynamic grounding of ensembles is not established: ensemble generation uses an SDE with heuristic noise (τ) and scheduler w(t); mapping samples to Boltzmann-weighted distributions, temperature dependence, and free-energy landscape fidelity are not validated.
- Calibration of confidence (pLDDT) is partial: correlation with LDDT-Cα is reported, but calibration quality (e.g., reliability curves, ECE/Brier scores), per-structure confidence, and ensemble-level uncertainty aggregation are not analyzed.
- Transferability of the pLDDT module to other models is claimed but untested: the idea is mentioned without empirical validation; systematic cross-model application and cross-domain calibration studies are missing.
- Architectural ablations are absent: the paper does not isolate contributions of adaptive layers, axial 4D RoPE, local attention masks, atom–residue grouping/ungrouping, or QK-norm/SwiGLU; targeted ablation studies are needed to identify essential components.
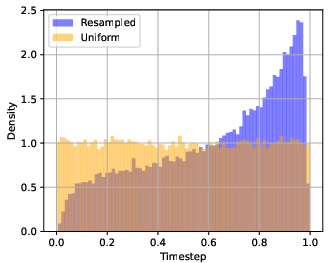
- Impact of timestep resampling and LDDT weighting schedule is underexplored: choices (e.g., oversampling near t=1, α(t) schedule) are justified intuitively but lack sensitivity analyses, ablations, or task-dependent tuning studies.
- Effect of using reference conformers in positional encoding is unclear: 4D axial RoPE uses “reference conformers” from rule-based cheminformatics; how much performance depends on this prior, and whether it introduces biases or limits generalization, is not quantified.
- Conditioning on ESM2-3B is fixed and frozen: the effect of end-to-end fine-tuning, alternative PLMs (size/domain), and multimodal conditioning (e.g., templates, predicted secondary structure, contact maps) is unexplored.
- No template usage experiments: while eschewing MSA is a design choice, experiments combining SimpleFold with template retrieval (without MSAs) could test performance ceilings and hybrid strategies.
- Training efficiency and compute costs are not reported: the paper highlights consumer-level inference but does not quantify training resources (FLOPs, GPU-days), scalability bottlenecks, or reproducibility constraints.
- Robustness and failure modes are not characterized: stress tests (e.g., disordered regions, repeats, membrane proteins, coiled-coils, metal-binding sites, non-canonical residues/PTMs) and failure case analyses are absent.
- CASP/CAMEO evaluation focuses on top-1 predictions; generative inference settings are narrow: ODE vs SDE sampling, τ/w(t) sweeps, and selection strategies (e.g., best-of-N, confidence-guided reranking) are not systematically studied for benchmark fairness.
- Interpreting why flow-tuned baselines underperform is speculative: the paper attributes ESMFlow/AlphaFlow drops to benchmark determinism, but does not test controlled setups (e.g., deterministic ODE sampling) to disentangle objective vs architecture effects.
- Downstream utility of ensembles in drug discovery is not evidenced: while ATLAS metrics improve, practical endpoints (cryptic pocket discovery, docking improvements, MD seeding quality, experimental validation) are not demonstrated.
- Extension beyond proteins is open: handling of nucleic acids, glycans, cofactors, prosthetic groups, and heteroatoms (beyond standard protein heavy atoms) is not supported or evaluated.
- Data quality vs quantity trade-offs are not analyzed: scaling data from ~2M to ~9M improves performance, but how label quality (experimental vs distilled), diversity, and redundancy affect learning is unknown.
- Post-hoc refinement and hybrid pipelines are not explored: integrating SimpleFold predictions with physics-based refinement, Rosetta-style optimization, or AF2 refinement could improve atomic detail; no experiments are provided.
Practical Applications
Overview
This paper introduces SimpleFold, a generative, flow-matching protein folding model that uses only general-purpose transformer blocks (no MSA, pair representations, or triangle updates). It generates full-atom structures, produces ensembles of conformations, includes a pLDDT confidence module, scales to 3B parameters, and achieves competitive accuracy on CASP14 and CAMEO22 while enabling efficient inference on consumer hardware. The practical applications below are derived from these findings, with sector links, deployment pathways, and feasibility notes.
Immediate Applications
Below are applications that can be deployed now, based on the model’s demonstrated capabilities, released code, and performance.
- Healthcare/Pharma: High-throughput single-chain structure prediction for drug discovery and target triage
- Use case: Rapid structure inference for novel or orphan proteins without MSA, supporting hit triage, target deconvolution, and mechanism-of-action hypotheses.
- Workflow: Sequence ingestion → ESM2 embedding → SimpleFold inference (full-atom) → pLDDT scoring → docking/virtual screening → optional energy minimization.
- Tools/products: “SimpleFold Server” or “Edge Folding” on lab workstations (M2 Max class machines), a Python SDK/API to batch-fold sequences, a QC module leveraging pLDDT.
- Assumptions/dependencies: Quality depends on PLM embeddings (ESM2-3B), single-chain focus (no complexes), sequence length limits (pretrain 256, finetune 512), side-chain accuracy adequate for docking with post-processing; experimental validation recommended.
- Biotech/Protein Engineering: Mutation impact assessment via ensemble generation
- Use case: Compare structural ensembles across variants to assess stability changes, loop flexibility, and cryptic pocket emergence relevant to enzyme activity and stability.
- Workflow: Variant enumeration → SimpleFold ensemble generation (tuning τ for stochasticity) → flexibility and contact profile comparison → prioritize variants for wet-lab assays.
- Tools/products: “Ensemble Explorer” that automates multi-variant folding and produces per-residue flexibility maps; reports that align with MD-derived metrics.
- Assumptions/dependencies: Ensemble quality improves with MD finetuning; use pLDDT to triage low-confidence regions; post hoc refinement (e.g., minimization) may be needed.
- Software/Data Platforms: Metagenomic sequence annotation at scale without MSA
- Use case: Annotate large sequence collections with predicted structures and confidence scores for downstream function prediction and remote homology classification.
- Workflow: Batch folding on CPU/GPU fleets → structure indexing → embeddings for similarity search; integrate with existing ML bioinformatics pipelines.
- Tools/products: Cloud-native folding microservice, structure database with pLDDT, automated retry and filtering based on confidence and structural sanity checks.
- Assumptions/dependencies: Distilled training data biases influence predictions; ensure compute quotas; design for long sequences and rare folds.
- Education/Training: Accessible on-device protein folding labs
- Use case: Undergraduate/graduate courses run local experiments to learn folding, ensemble generation, and confidence assessment without a large server cluster.
- Workflow: Notebook-based exercises reproducing CASP/CAMEO-style evaluation; explore τ effects on ensembles; visualize pLDDT vs. LDDT relationships.
- Tools/products: Teaching notebooks, visualization plugins, lab curricula that integrate SimpleFold outputs with PyMOL/VMD.
- Assumptions/dependencies: Requires modern laptop with ample memory; curated datasets to avoid dual-use risks; student-friendly wrappers.
- Structural QC: Confidence estimation and triage using pLDDT
- Use case: Rank and filter predicted structures for downstream pipelines (docking, design, annotation) using per-residue pLDDT.
- Workflow: Fold → pLDDT inference → thresholding per region → route high-confidence structures for further tasks and flag uncertain regions for caution.
- Tools/products: “Confidence Filter” service (pLDDT-only module) that can score structures from SimpleFold or other models.
- Assumptions/dependencies: Reported Pearson correlation (~0.77 with LDDT-Cα); use caution for flexible loops; calibrate thresholds per application.
- MD Cost Reduction: Approximate dynamics via generative ensembles
- Use case: Reduce reliance on costly MD by generating ensembles that qualitatively capture flexibility, contact variability, and exposure metrics.
- Workflow: Ensemble generation → metrics (RMSF, contacts, PCA) → identify flexible regions or transient pockets → selective MD for top targets.
- Tools/products: “MD-Lite” pipeline that benchmarks ensembles against ATLAS-style metrics; integrates with docking to account for induced fit.
- Assumptions/dependencies: Best results with MD finetuning; ensembles approximate, not replace, MD; validation of critical decisions via experiments or physics-based simulations.
- Operations/Cost: Democratized compute for small labs and startups
- Use case: Run competitive folding locally, reducing cloud and MSA costs, enabling faster iteration cycles.
- Workflow: On-device inference → local cache of folded structures → lightweight CI/CD for data science workflows.
- Tools/products: Dockerized folding service; model size selection (100M–3B) based on device resources; batching and caching utilities.
- Assumptions/dependencies: Hardware requirements (e.g., M2 Max 64GB); throughput planning; ensure licensing compliance for PLM and datasets.
Long-Term Applications
Below are applications likely to require further research, scaling, integration, or regulatory consideration.
- Healthcare/Clinical Genomics: Variant pathogenicity prediction integrating structure and dynamics
- Use case: Clinical interpretation workflows that use SimpleFold ensembles to predict structural disruption, altered dynamics, and functional impact.
- Workflow: Genomic variant calling → ensemble folding → feature extraction (stability changes, contact disruptions, cryptic pocket formation) → classifier for pathogenicity.
- Tools/products: “Structure-informed Variant Interpreter.”
- Assumptions/dependencies: Requires large labeled datasets, calibrated clinical models, robust generalization, regulatory validation; current single-chain focus may limit scope.
- Drug Discovery: Ensemble-aware structure-based design and docking at scale
- Use case: Dock ligands against diverse conformational states from SimpleFold ensembles to better capture induced-fit and cryptic pocket binding.
- Workflow: Generate ensembles → ensemble docking/scoring → consensus ranking → design-make-test cycle; integration with generative molecule design.
- Tools/products: “Ensemble Docking Engine” integrated with medicinal chemistry tools.
- Assumptions/dependencies: Needs validated scoring functions for ensembles; side-chain and pocket fidelity; often requires post-processing (minimization/MD); complex targets may need future multi-chain support.
- Complexes and Interactions: Extension of SimpleFold to multi-chain assemblies and biomolecular interactions
- Use case: Predict structures of complexes (protein–protein, protein–nucleic acid), enabling systems-level design.
- Workflow: Multi-chain conditioning → generative ensemble of complex states → interface analysis and affinity estimation.
- Tools/products: “SimpleFold-Complex” with interaction-aware conditioning and sampling.
- Assumptions/dependencies: Architectural extensions (pairwise/context modeling) or learned interaction tokens; new datasets; physics-informed constraints.
- Synthetic Biology/Materials: Closed-loop sequence→structure→function optimization
- Use case: Automated enzyme and protein material design leveraging rapid folding and ensemble feedback to navigate sequence space efficiently.
- Workflow: Generative sequence design → SimpleFold folding and ensemble metrics → objective-driven optimization → wet-lab validation → iterate.
- Tools/products: “Autonomous Protein Design Studio.”
- Assumptions/dependencies: Requires robust surrogate function models; multi-objective optimization; strong lab integration; risk management for dual-use concerns.
- Environmental/Agriculture/Energy: Discovery of enzymes for bioremediation, crop traits, and biofuel production
- Use case: Screen metagenomic sequences to identify promising enzymes with desired structure/dynamics properties (e.g., substrate-binding flexibility).
- Workflow: High-throughput folding and ensemble screening → candidate prioritization → characterization and directed evolution.
- Tools/products: “Enzyme Prospecting Pipeline.”
- Assumptions/dependencies: Requires functional assays; may need complex cofactor/ligand modeling; depends on sequence diversity and data quality.
- Structural Biology Tooling: Cryo-EM map fitting support via rapid per-chain modeling
- Use case: Provide initial chain models or ensemble priors to fit into density maps during early-stage reconstruction.
- Workflow: Chain sequence folding → initial fit → iterative refinement → ensemble-informed ambiguity resolution.
- Tools/products: “Cryo-EM Chain Builder” module.
- Assumptions/dependencies: Complexes and flexible regions are challenging; coupling to map-fitting algorithms; experimental constraints needed.
- Policy/Governance: Compute and access governance for generative bio models
- Use case: Frameworks for model access, guardrails, monitoring, and safe use—balancing democratization with dual-use risk mitigation.
- Workflow: Risk assessment → tiered access policies → audit trails → non-proliferation guidelines for generative bio tools.
- Tools/products: “BioML Governance Toolkit.”
- Assumptions/dependencies: Multistakeholder alignment; evolving regulatory norms; transparent reporting on training data and capabilities.
- Standards and Benchmarking: Ensemble-aware evaluation standards
- Use case: Community standards that go beyond single ground-truth targets to ensemble-aware metrics (e.g., RMSF, transient contacts, PCA distributions).
- Workflow: Benchmark panel design → shared datasets → agreed metrics and protocols → public leaderboards.
- Tools/products: “Ensemble Benchmarks” akin to ATLAS-style evaluations.
- Assumptions/dependencies: Consensus-building; curation of high-quality data; reproducibility infrastructure.
- Edge/Personal Biocomputing: Consumer-grade biodesign tools with safety overlays
- Use case: Empower small labs/citizens to explore protein folding and engineering responsibly on local devices.
- Workflow: On-device folding; integrated safety prompts and usage boundaries; community education resources.
- Tools/products: “Safe BioDesign Desktop.”
- Assumptions/dependencies: Strong safety and ethics program; curated datasets; limited feature sets to avoid misuse.
Cross-cutting Feasibility Notes
- Model scope: Current SimpleFold focuses on single-chain, full-atom folding; complex assembly support is a future extension.
- Data dependence: Trained on large distilled datasets (AFDB/AFESM) and PDB; biases and quality thresholds (pLDDT filters) affect generalization.
- Confidence: pLDDT correlates with LDDT-Cα; use per-residue confidence for triaging decisions; flexible loops often have lower confidence.
- Ensembles: τ tuning affects diversity; MD finetuning improves ensemble fidelity; ensembles are approximations—validate critical steps via physics-based methods.
- Compute: Efficient inference on consumer hardware is possible; throughput and sequence length constraints must be planned; model size choice matters.
- Integration: Downstream tasks (docking, design, function prediction) often need post-processing (energy minimization/clash resolution) and experimental validation.
- Safety and policy: Generative bio capabilities call for access control, monitoring, and educational guardrails, especially as on-device use scales.
Glossary
- AdamW optimizer: An optimization algorithm that decouples weight decay from the gradient update to improve training stability. "We use the AdamW optimizer~\citep{loshchilov2019decoupled} with learning rate $0.0001$"
- Adaptive layers: Transformer layers modulated by conditioning variables (e.g., the flow timestep) to enable time-dependent behavior. "employs standard transformer blocks with adaptive layers"
- AFDB (AlphaFold Protein Structure Database): A large public database of protein structures predicted by AlphaFold. "Additionally, we use the SwissProt set from AFDB."
- AFESM: A combined resource that merges AFDB and the ESM Metagenomic Atlas into clustered distilled protein structure sets. "AFESM~\citep{yeo2025afesm} further combines these two resources and categorizes all distilled structures from both datasets into 5.12M non-singleton structural clusters."
- AFESM-E: An extended version of AFESM that includes additional members per cluster to increase data scale. "we explore an extended version AFESM (which we call AFESM-E)"
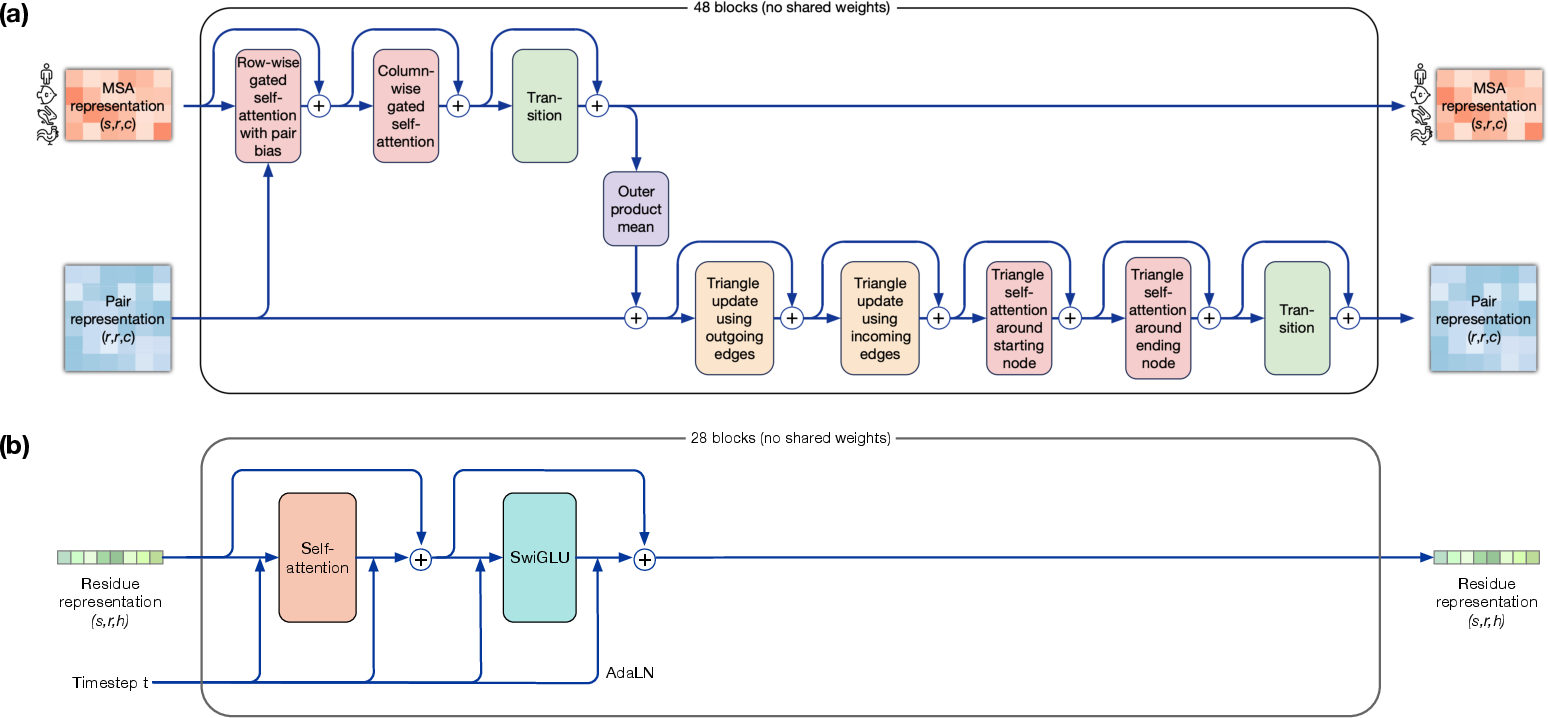
- AlphaFold2: A state-of-the-art protein folding system that uses MSA, pair representations, and triangular modules. "AlphaFold2 introduced domain specific network modules like triangle attention and design decisions like explicitly modeling interactions between single and pair representations."
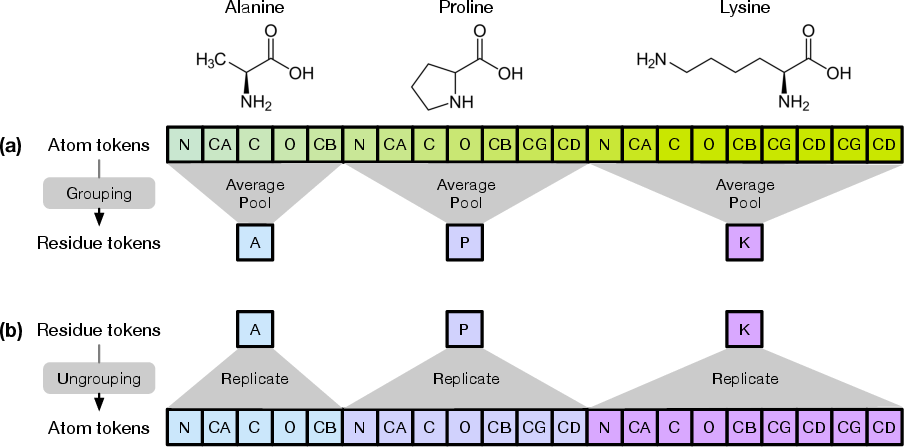
- Amino acid (AA) sequence: The linear chain of amino acids that specifies a protein; the input condition for folding models. "from its amino acid (AA) sequence"
- ATLAS: A benchmark dataset comprising molecular dynamics (MD) simulation ensembles used to evaluate ensemble generation. "ATLAS contains contains all-atom MD simulations of 1390 proteins."
- Axial RoPE (4D axial RoPE): Applying rotary position embeddings along multiple axes (e.g., 3D coordinates and residue index) to encode multidimensional structure. "we extend the positional embedding to a 4D axial RoPE."
- CAMEO22: A benchmark suite for evaluating protein structure prediction accuracy on diverse targets. "We evaluate {SimpleFold} on two widely adopted protein structure prediction benchmarks: CAMEO22 and CASP14"
- CASP14: A blind assessment benchmark for protein structure prediction, emphasizing challenging targets. "On the more challenging CASP14 benchmark, {SimpleFold} achieves even better performance than ESMFold."
- Cheminformatic method: A rule-based approach from cheminformatics to construct local atomic conformers. "predicted at the amino acid level by a rule-based cheminformatic method."
- Distilled structures: Computationally predicted protein structures used as training data via distillation rather than direct experimental measurement. "train it on approximately 9M distilled protein structures together with experimental PDB data."
- Equivariant architectures: Neural network designs that preserve geometric symmetries (e.g., rotations) by construction. "rely on equivariant architectures to generate physically meaningful results"
- ESM2-3B (Protein LLM): A 3B-parameter protein LLM used to embed sequences into residue-level features. "We leverage ESM2-3B ~\citep{lin2023evolutionary} in all our models to encode the AA sequence"
- Euler step: A single explicit integration step used to approximate the solution of a differential equation. "one step Euler"
- Euler–Maruyama integrator: A numerical scheme for integrating stochastic differential equations. "We apply the EulerâMaruyama integrator~\citep{sit}:"
- Exponential moving average (EMA): A smoothing technique that maintains a decayed running average of model parameters. "we apply an exponential moving average (EMA) of all model weights with a decay of $0.999$"
- Flow matching: A generative modeling paradigm that learns a time-dependent velocity field to transform noise into data via an ODE. "Flow-matching generative models ~\citep{lipman2023flow,albergo2023building} approach generation as a time-dependent process"
- Fourier positional embeddings: Sinusoidal encodings used to represent continuous positions (e.g., coordinates) for transformer inputs. "is encoded through Fourier positional embeddings."
- GDT-TS: Global Distance Test–Total Score; a measure of overall structural similarity between two protein models. "TM-score and GDT-TS assess global structural similarity"
- Gibbs free energy: A thermodynamic quantity minimized by native protein structures; used to motivate generative modeling of ensembles. "as a non-deterministic minimizer of the the Gibbs free energy of the atomic system."
- Langevin-style SDE: A stochastic differential equation combining deterministic drift with noise to sample from learned distributions. "We perform stochastic generation using a Langevin-style SDE formulation of the flow process"
- LDDT (Local Distance Difference Test): A local accuracy metric measuring differences in inter-atomic distances between predicted and true structures. "We also include an additional local distance difference test (LDDT) loss"
- Logistic-normal distribution: A distribution on (0,1) obtained by transforming a normal variable through the logistic function; used to sample timesteps. "where is logistic-normal distribution~\citep{atchison1980logistic}"
- Molecular dynamics (MD): Physics-based simulations of atomic motion used to generate ensembles of protein conformations. "ATLAS contains contains all-atom MD simulations of 1390 proteins."
- Multiple sequence alignment (MSA): An alignment of evolutionarily related sequences used to extract coevolutionary signals for folding. "multiple sequence alignments (MSAs) of AA sequences"
- MSA subsampling: A procedure that samples subsets of MSA entries to induce stochasticity in deterministic predictors. "MSA subsampling introduces more stochasticity to AlphaFold2"
- Orphan proteins: Proteins with few or no homologs, for which MSA-based methods are less effective. "for orphan proteins (those with few or no close homologs)"
- Ordinary differential equation (ODE): A differential equation describing the deterministic time evolution used in flow matching. "integrating an ordinary differential equation (ODE) over time."
- Pair representations: Explicit pairwise feature tensors modeling interactions between residue pairs. "explicit pair representations"
- pLDDT: Predicted LDDT; a per-residue confidence score indicating estimated local accuracy. "we develop an additional predicted LDDT (pLDDT) module"
- PDB (Protein Data Bank): The primary repository of experimentally determined biomolecular structures. "we include around 160K structures from PDB"
- PLM (Protein LLM): A neural model trained on protein sequences to learn contextual residue embeddings. "protein LLMs (PLM) tend to outperform approaches that rely on MSA"
- Pushforward operator: The transformation of a probability distribution under a mapping; used to relate base and target distributions. "where denotes the pushforward operator."
- QK-normalization: A technique that normalizes the query and key vectors in attention to improve stability and performance. "including QK-normalization~\citep{sd3}"
- Rectified flow: A specific linear-interpolant formulation of flow matching equivalent to diffusion under Gaussian marginals. "(also referred to as a rectified flow \citep{liu2022flow, sd3})"
- Residue trunk: The main transformer module operating at the residue level that carries most of the model capacity. "The residue trunk contains most of the parameters of the model"
- Reverse-time Wiener process: Brownian motion run backward in time, appearing in the stochastic flow sampling formulation. "is a reverse-time Wiener process"
- RoPE (Rotary position embedding): A positional encoding method that injects relative position via complex rotations in attention. "we employ rotary position embedding (RoPE)~\citep{su2024roformer}"
- Side chain: The set of atoms extending from the backbone that differentiates amino acids chemically. "side chain atoms"
- Signal-to-noise ratio (SNR): The ratio controlling relative strength of signal to noise; used to schedule stochasticity. "following SNR of flow process"
- SO(3) data augmentation: Random 3D rotations applied to structures to enforce rotational invariance during training. "we apply SO(3) data augmentation during training"
- SwiGLU: A gated activation function used in transformer feedforward networks for improved performance. "and SwiGLU~\citep{shazeer2020glu} in place of standard FFN"
- SwissProt: A curated, high-quality subset of UniProt; its AFDB predictions are used as distilled training data. "we use the SwissProt set from AFDB."
- Timestep resampling: A strategy to sample training timesteps from a non-uniform distribution to emphasize certain parts of the flow. "Timestep resampling."
- TM-score: A length-independent measure of global structural similarity between protein models. "TM-score and GDT-TS assess global structural similarity"
- Triangle updates: Specialized pairwise geometry update modules used in AlphaFold-like architectures. "like pair representations and triangle updates"
Collections
Sign up for free to add this paper to one or more collections.

