- The paper introduces PairSAE, which extracts detailed, monosemantic features from both sequence and pairwise embeddings for improved interpretability.
- It employs N-mode SVD compression and a sparse autoencoder to tackle quadratic complexity while ensuring clear mechanistic insights in protein structures.
- PairSAE demonstrates strong binding affinity prediction via LASSO regression, outperforming standard models and providing actionable latent markers.
Mechanistic Interpretability of Pair Representations in Protein Co-Folding: The PairSAE Framework
Motivation and Context
Protein structure prediction models, particularly those leveraging pairformer architectures, have achieved substantial success in predicting biomolecular structure and interactions, but consistently lack transparent interpretability at the latent representation level. Traditional sparse autoencoders (SAEs) are effective in transformer-style models, but do not scale efficiently to pairwise representations, where a naive application obscures mechanistic concepts and results in quadratic feature complexity. The presented work introduces PairSAE, designed to extract interpretable, monosemantic token-level features from both sequence and pairwise embeddings in Boltz-2 representations. PairSAE addresses both technical scalability and conceptual alignment, offering insights into structural biology foundation models’ latent spaces.
Methodology
PairSAE operates by first compressing Boltz-2 pairwise tensors Z using N-mode SVD, yielding token-wise interaction summaries—mode-1 and mode-2 left singular vectors—which encapsulate both row and column roles in interaction matrices. These condensed embeddings are concatenated with sequence representations, normalized, and fed into a sparse autoencoder equipped with BatchTopK and ReLU nonlinearities. This shared latent feature set is decoded back into both sequence and pairwise spaces via linear projections, and trained using a robust Matryoshka SAE loss spanning nested dictionary sizes, plus an auxiliary loss for feature revitalization. The entire pipeline is optimized efficiently by Monte Carlo sampling over pairwise reconstructions, maintaining tractable scaling.
Interpretability Evaluation
PairSAE is evaluated on Boltz-2 activations from the PLINDER dataset, training two SAE instances on the third recycling step at layers 33 and 64. Mechanistic interpretability is assessed through linear probes targeting UniProt residue annotations and PLINDER system-level concepts. Each feature is normalized, and single-threshold classifiers are constructed by grid search to maximize F1 scores at both token and complex levels.
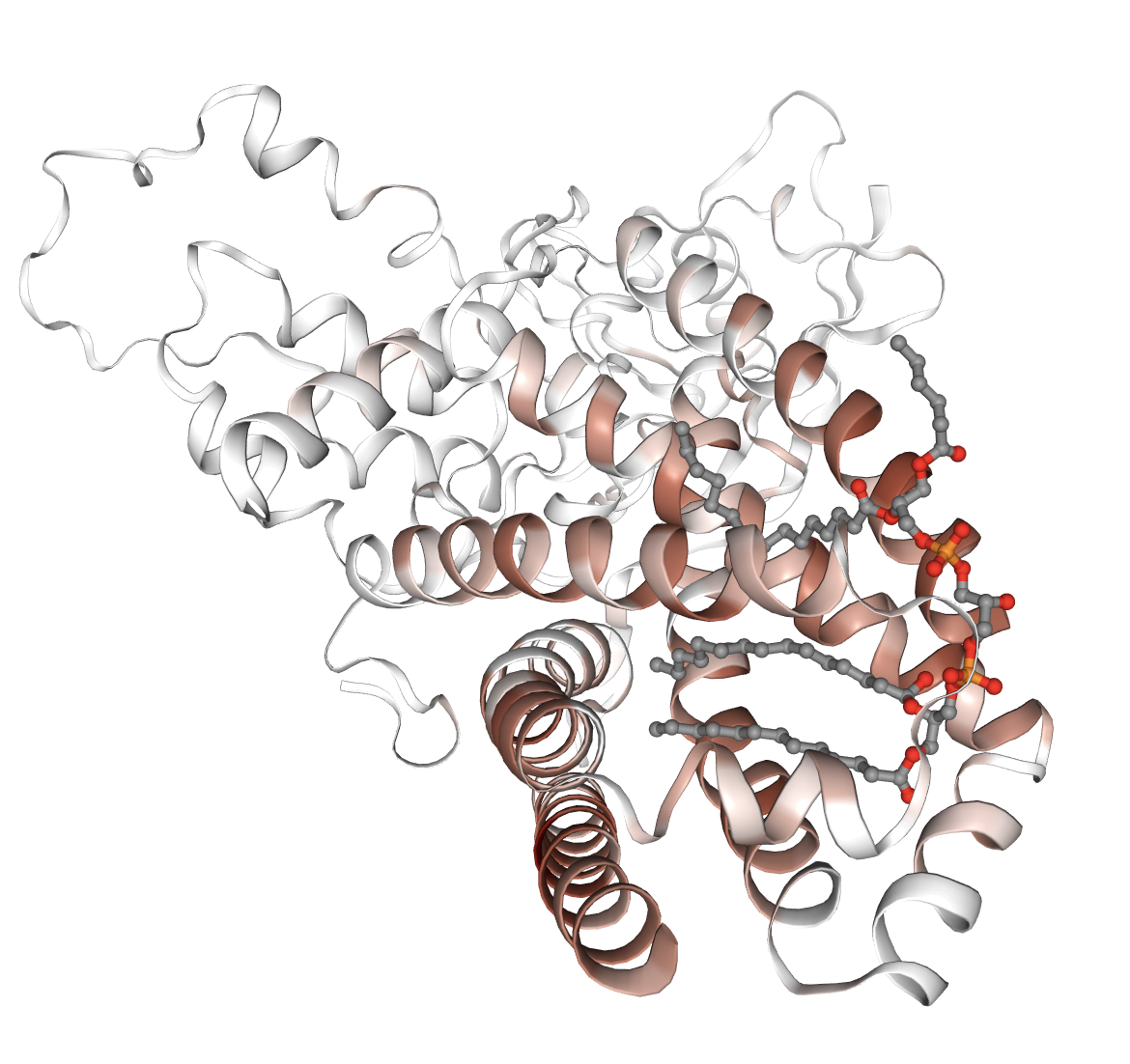
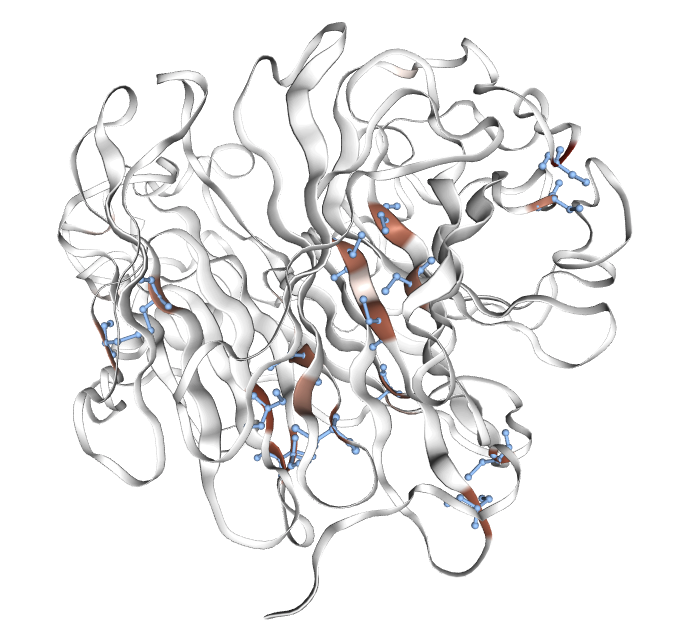
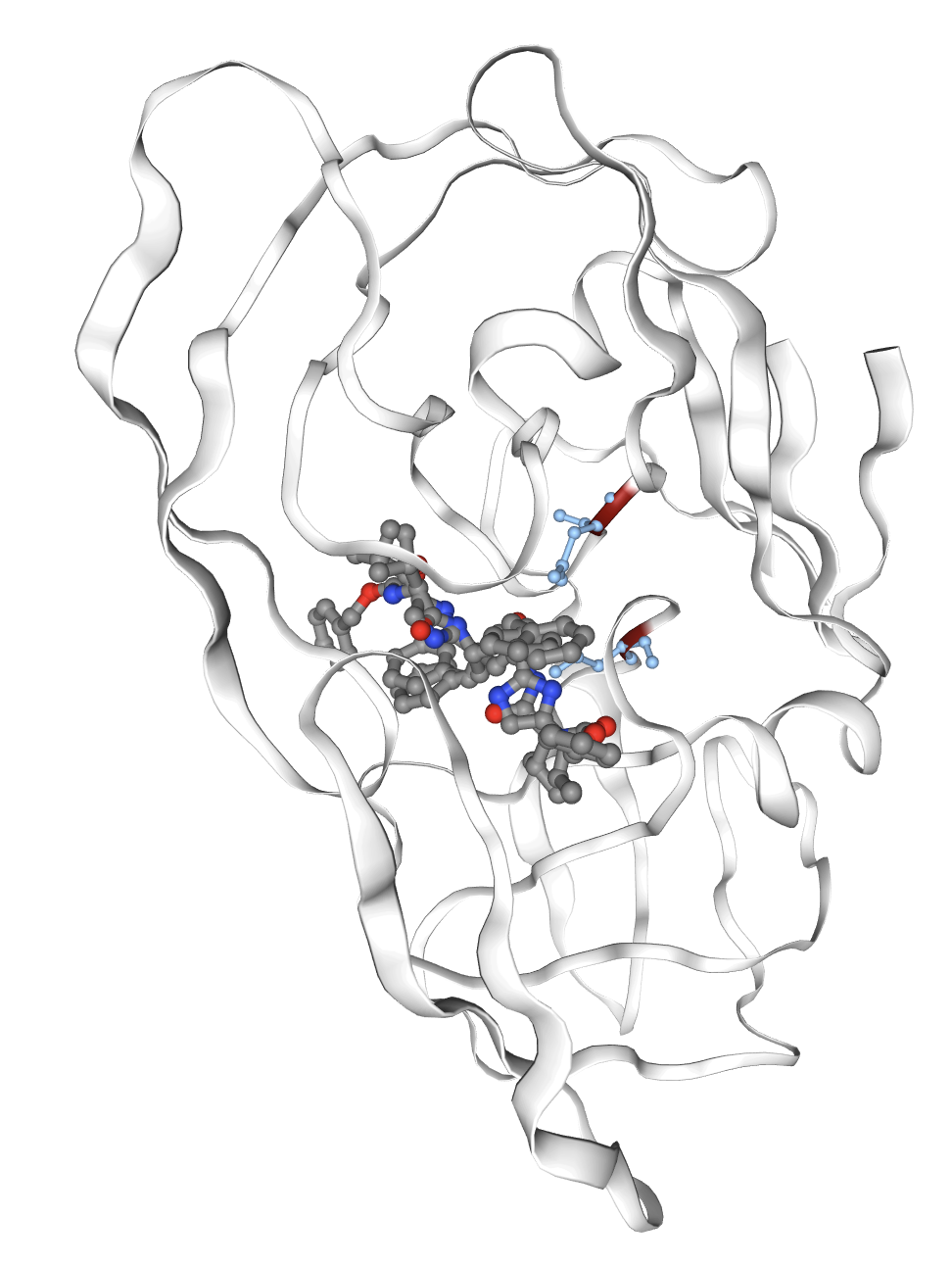

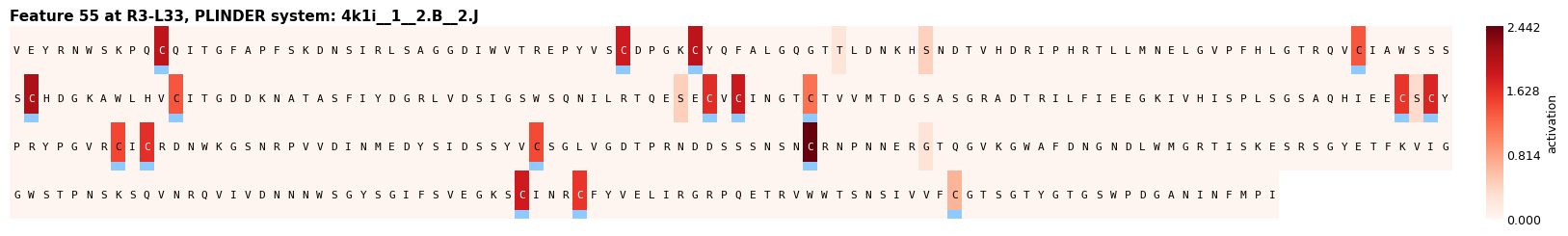

Figure 1: Examples of interpretable features extracted by PairSAE at two depths; features correspond to transmembrane domains, disulfide bonds, and protease active sites—all aligning with UniProt ground truth.
Strong conceptual alignment is observed: features extracted by PairSAE sharply correlate with functional annotations, outperforming models such as ESM2-650M whose last layer neurons yield substantially lower F1 scores. In particular, PairSAE captures 29.1% (R3-L32) and 24.0% (R3-L64) of concepts with token-level F1≥0.5, versus only 0.8% for ESM2-650M, and comparable gains are observed at the complex level.
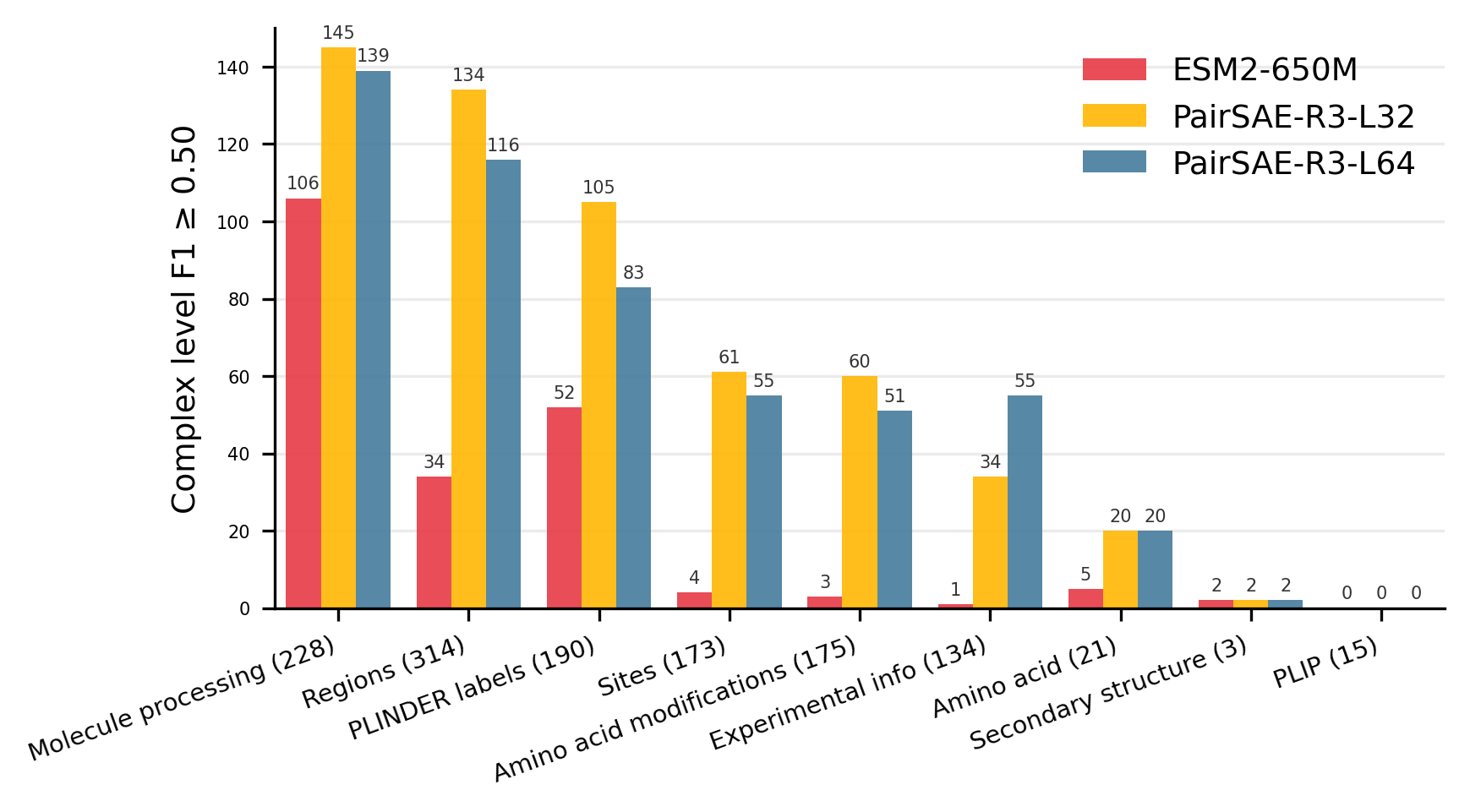
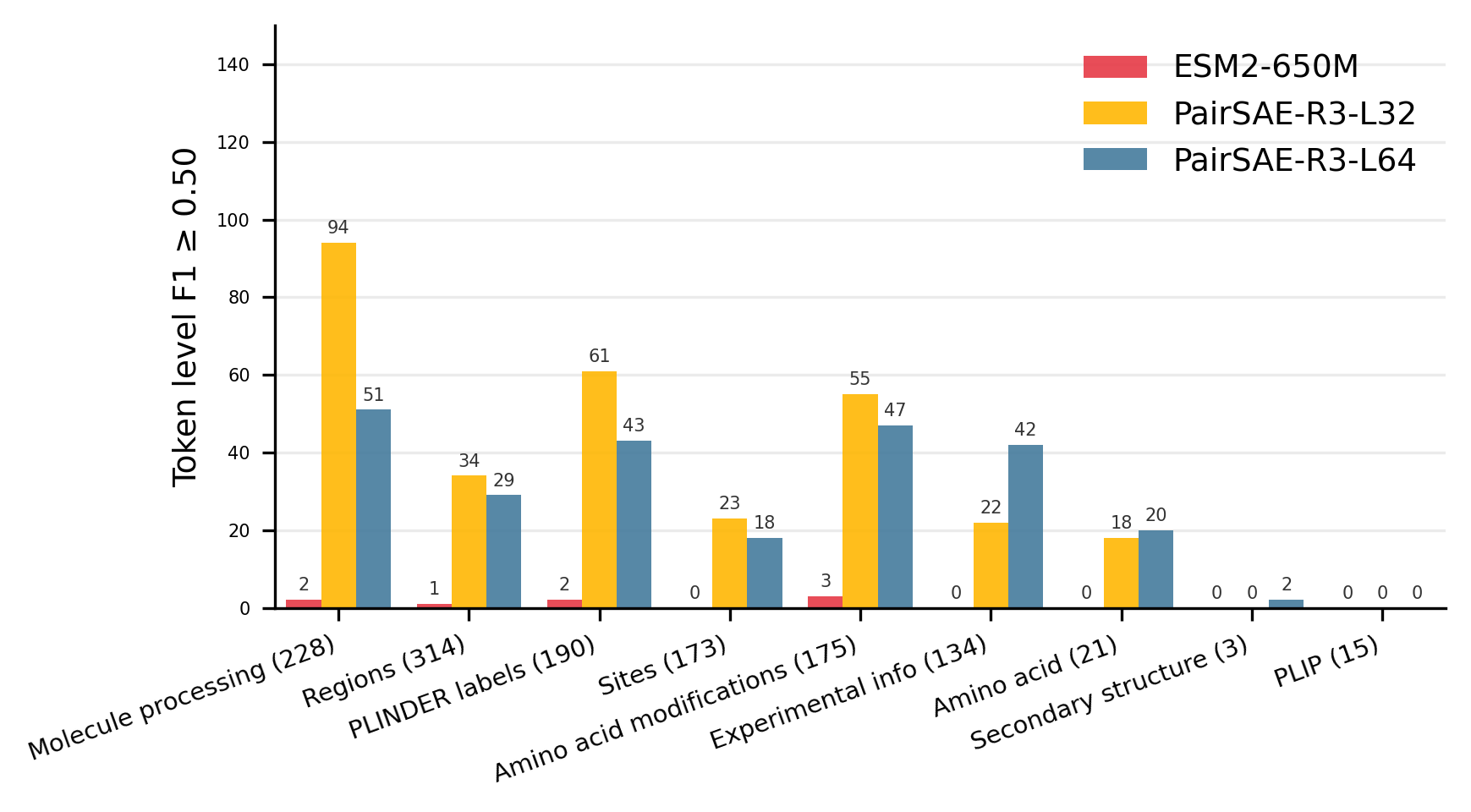
Figure 2: Distribution of concept counts with F1≥0.5, grouped by annotation category, using token and complex-level metrics.
Hypothesis Generation and Binding Affinity Prediction
Beyond annotation alignment, PairSAE features are leveraged for hypothesis generation regarding protein-ligand binding affinity, a critical downstream task. Max-pooled latent activations over the token dimension are used to form complex-level embeddings, which are subsequently regressed (via LASSO) against Boltz-2 predicted affinity values. The model achieves a test set R2 of 0.528 from the R3-L64 SAE using 291 sparse features out of 16,384, indicating strong predictive power and substantial model compression. Notably, most affinity-predictive features activate on ligands—highlighting the capacity for latent structural hypothesis extraction even in regions with limited annotation.
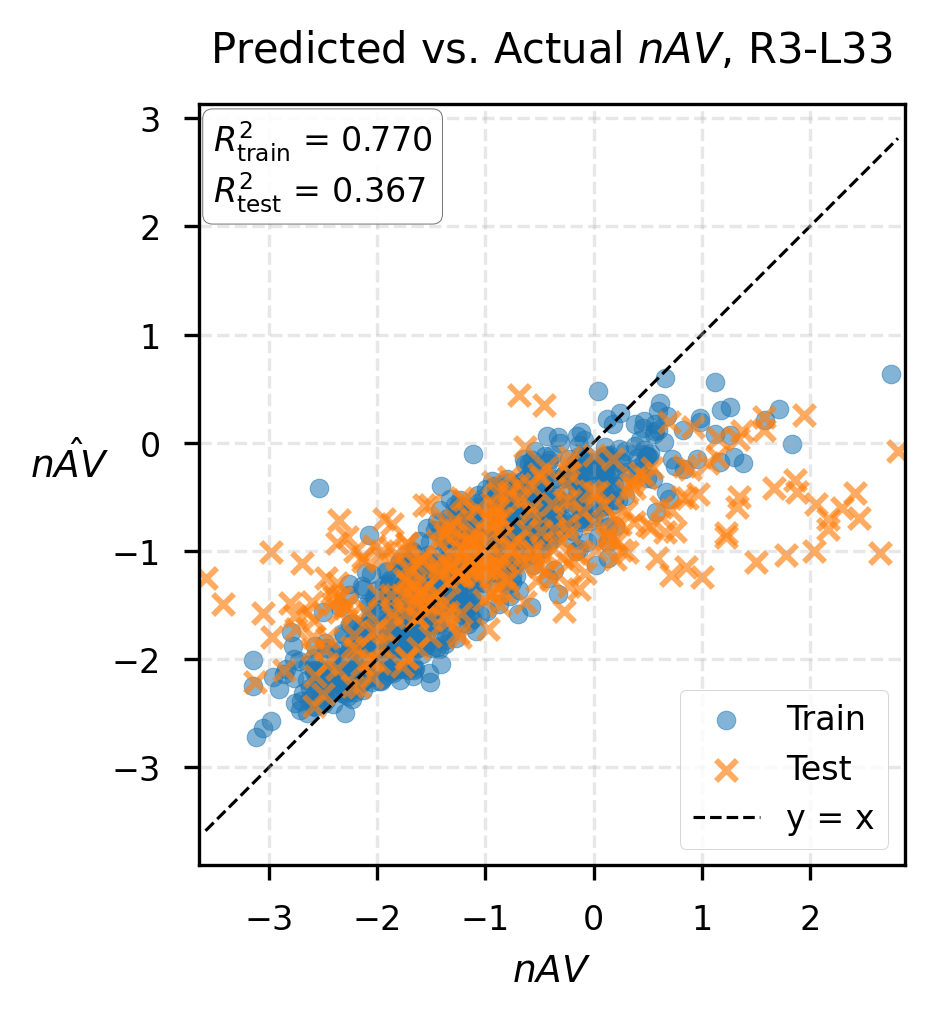
Figure 3: True vs. predicted Boltz-2 affinity values using PairSAE features (LASSO regression), revealing accurate recovery of high-affinity complexes.
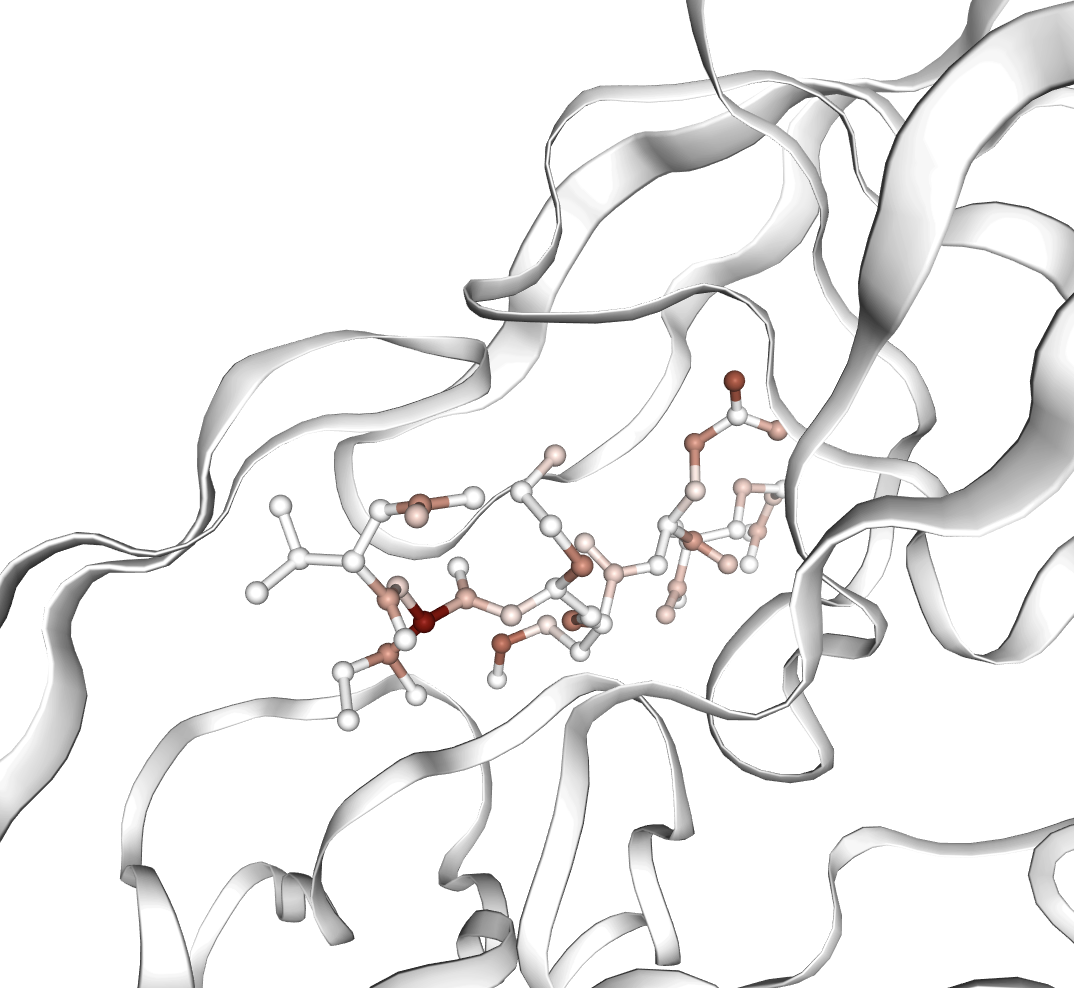
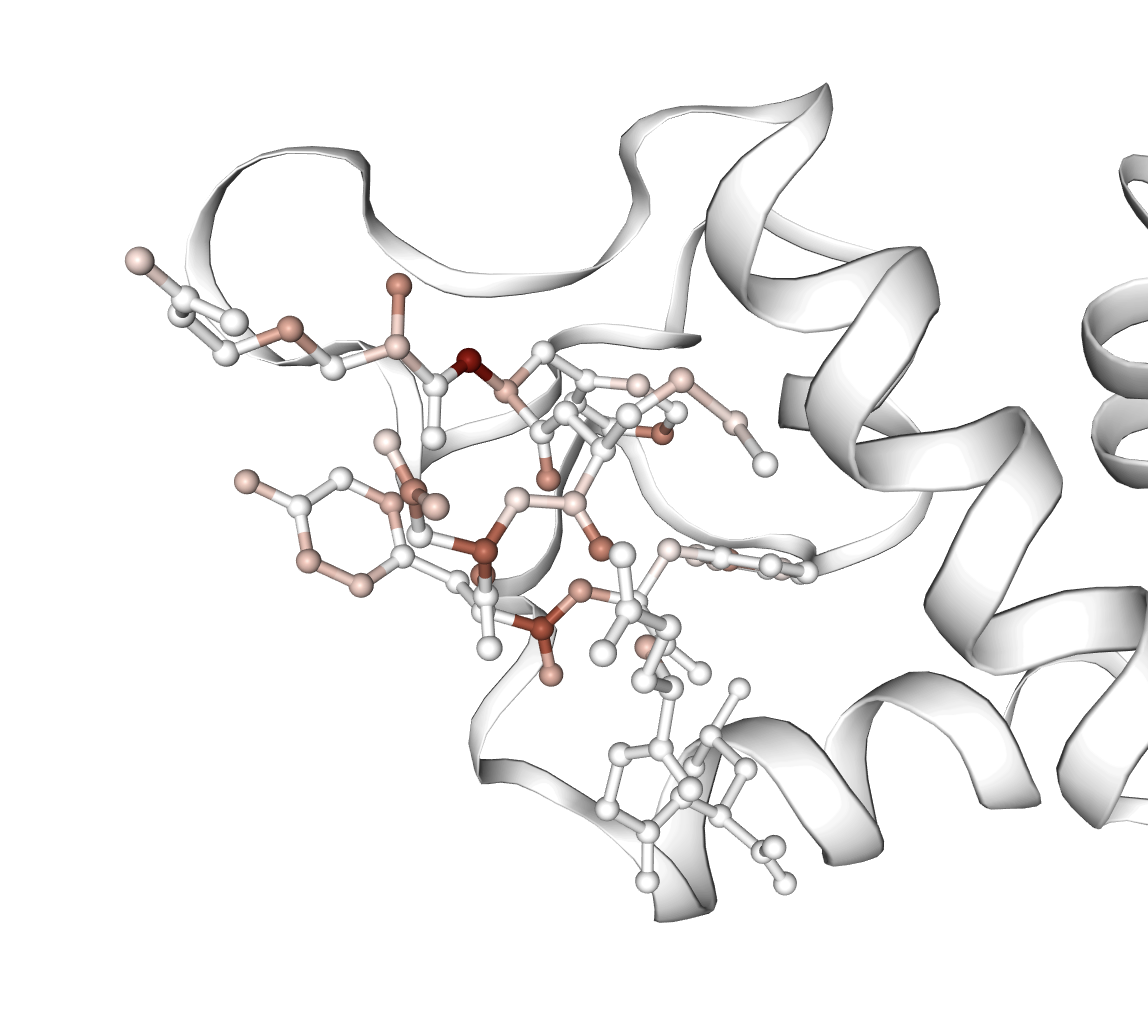
Figure 4: Visualization of ligands strongly activating important affinity-predictive features, demonstrating mechanistic localization.
Group difference analysis delineates interpretable latent markers for affinity-associated and non-affinity complexes. Certain features (e.g., R3-L64:2299, R3-L33:3888) are cleanly split by activation state, with substantial differences in predicted affinity and physically plausible docking behavior.
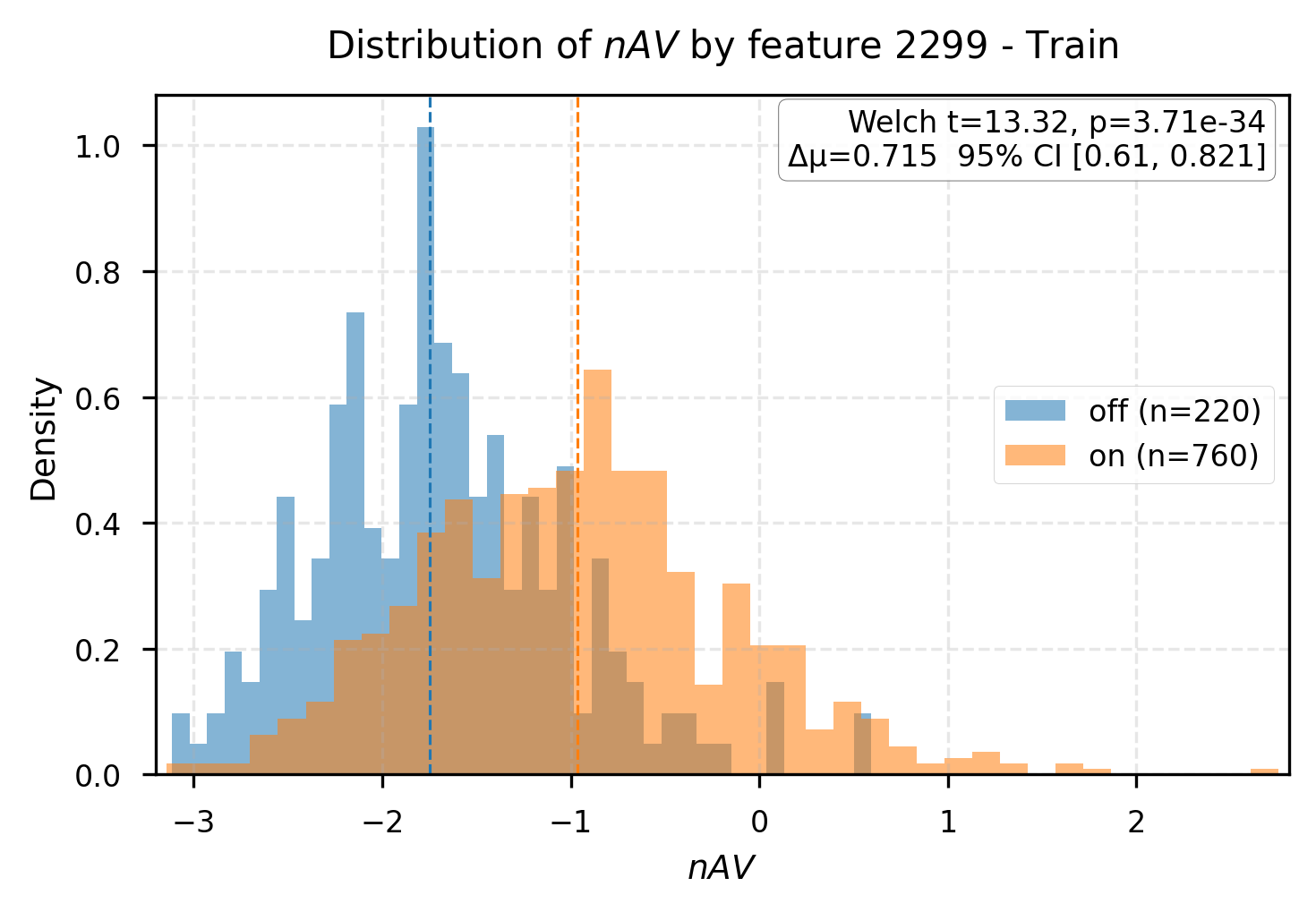
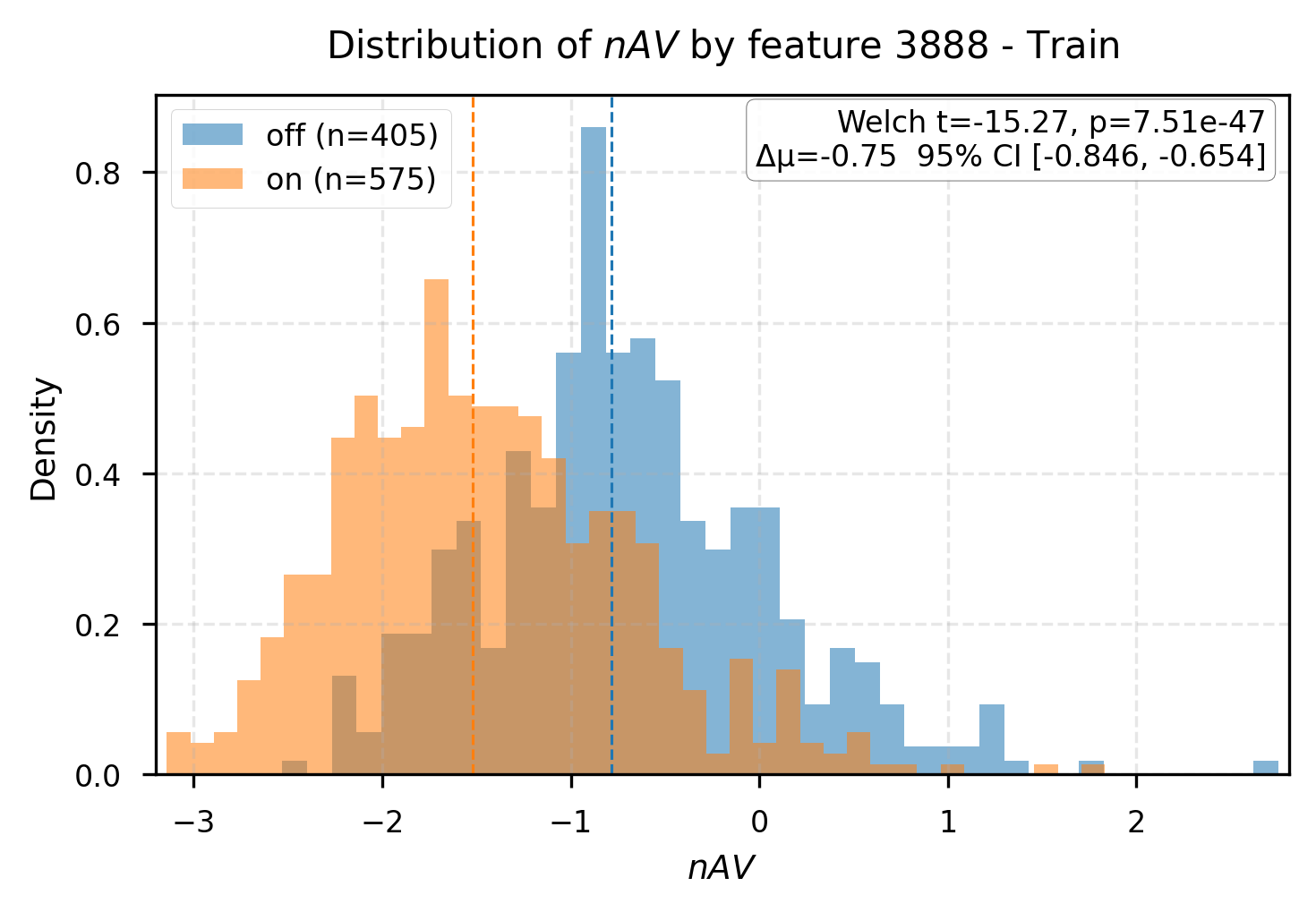
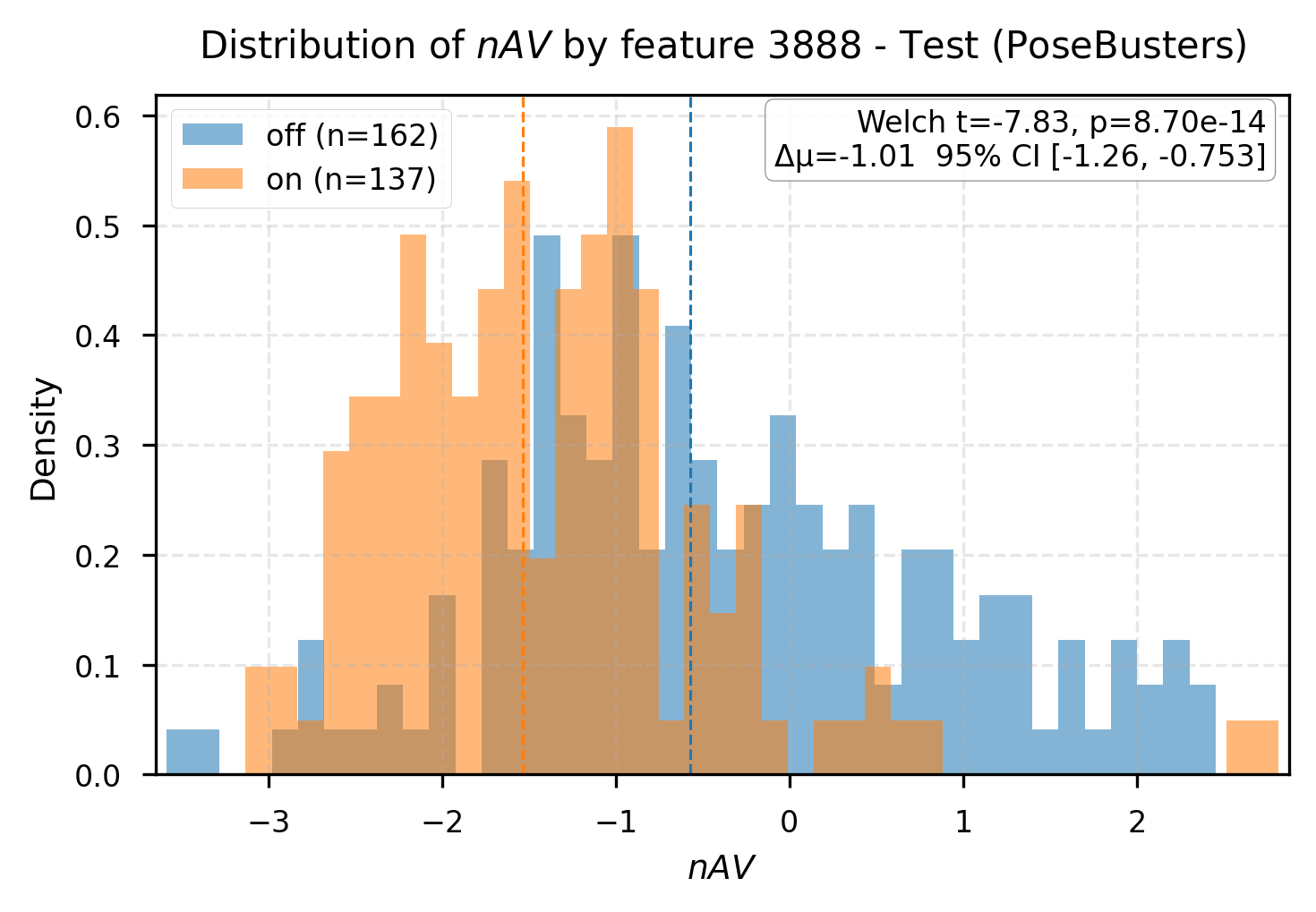
Figure 5: Group differences in affinity values for samples split by feature activation, mapping latent concepts to structural outcomes.
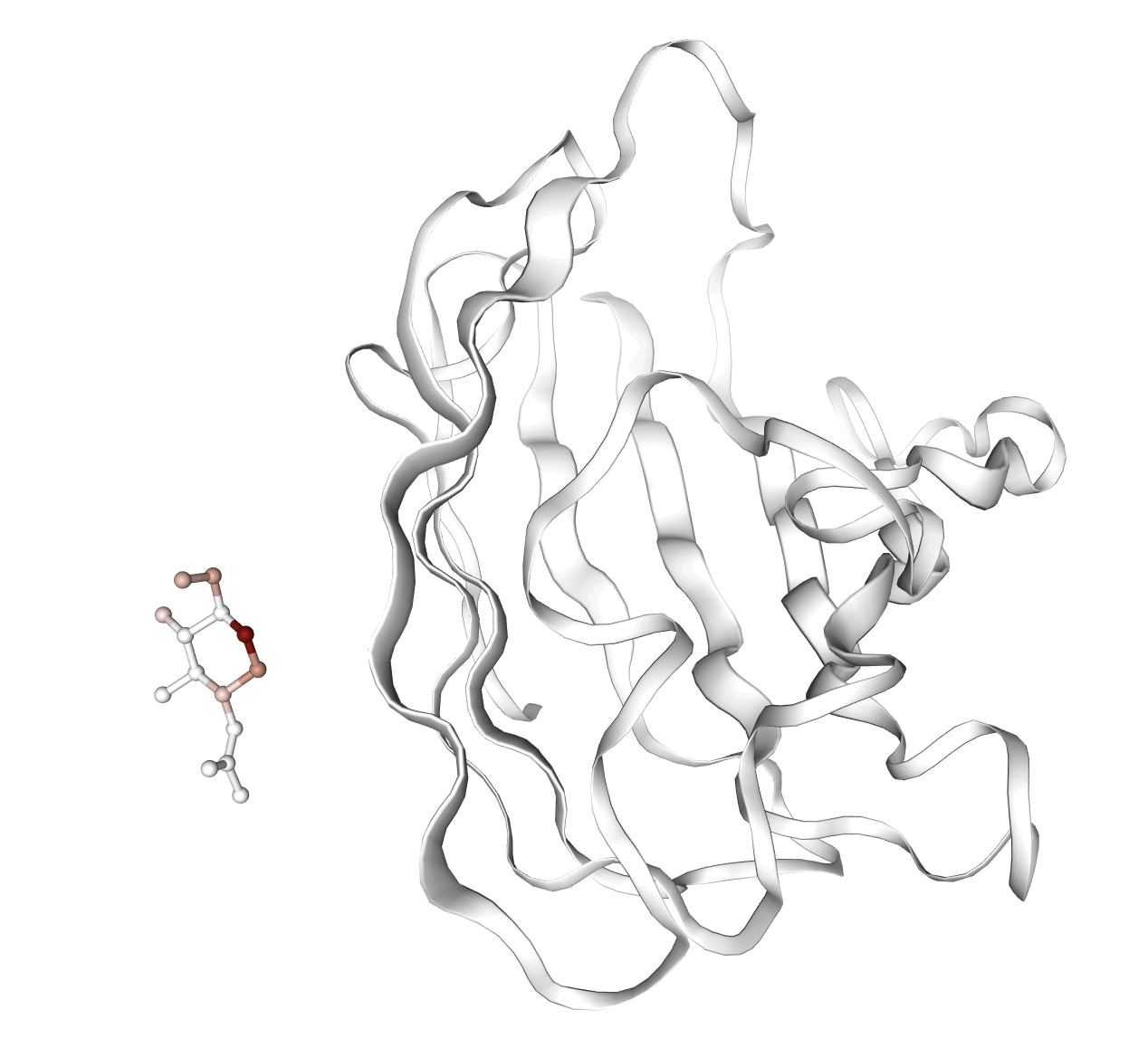
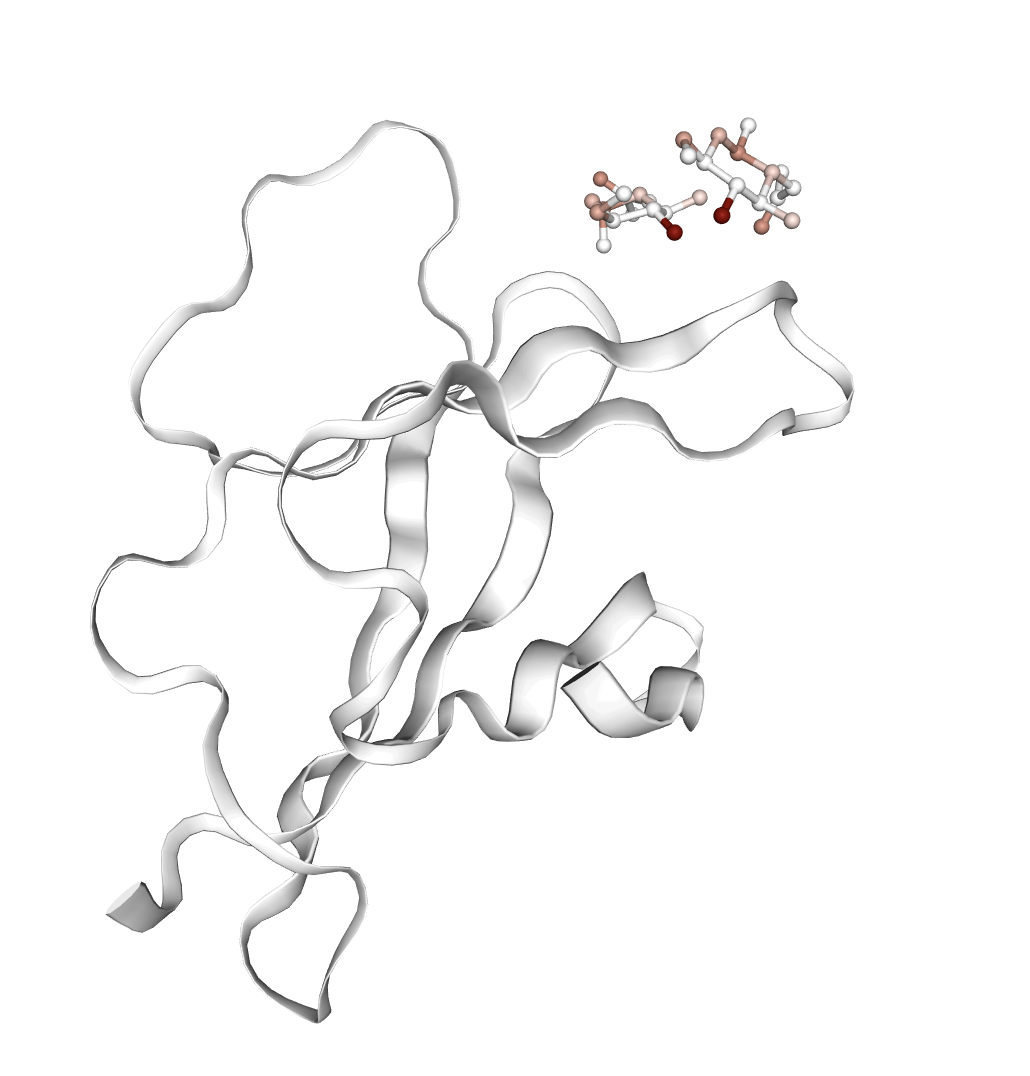
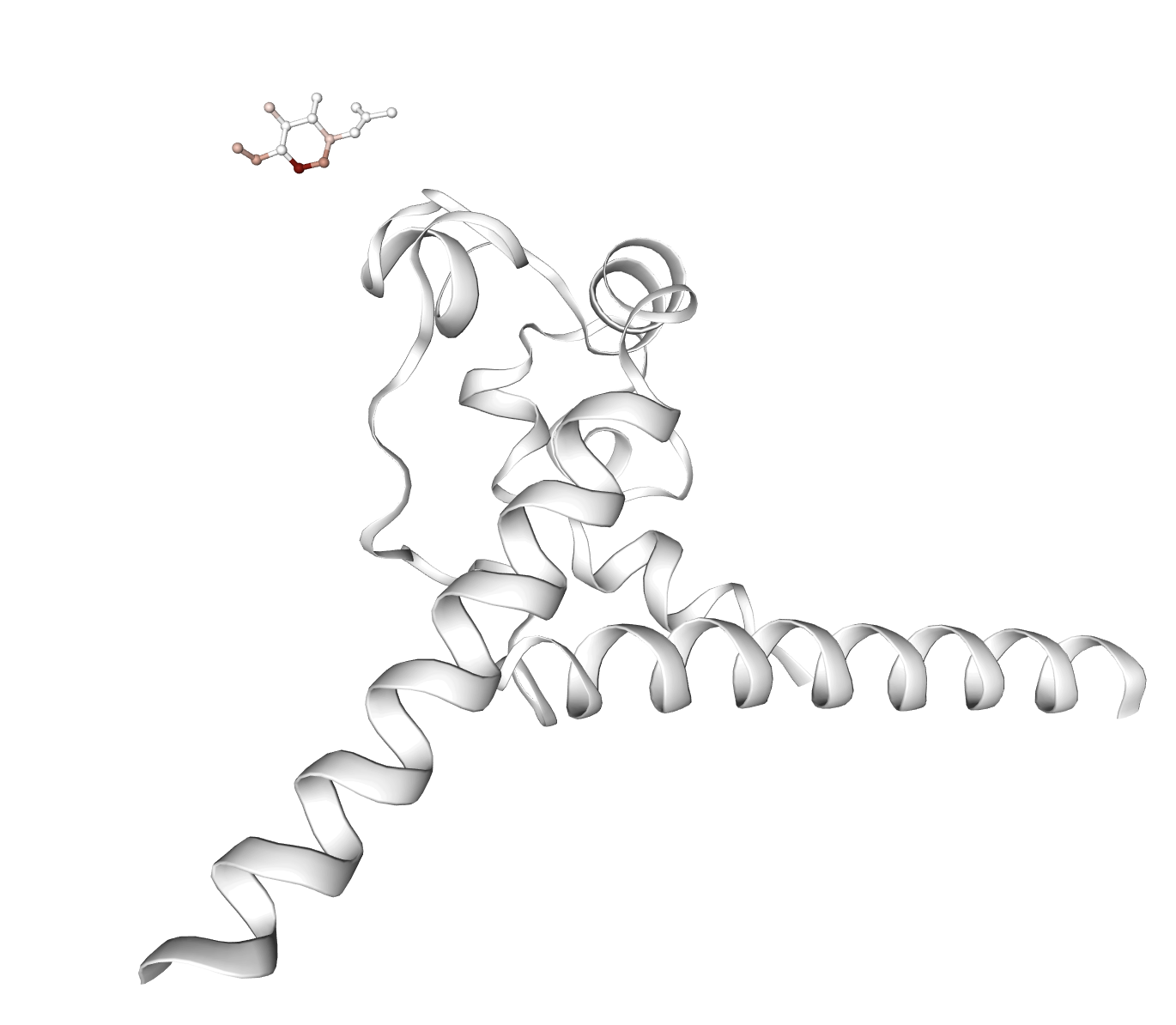
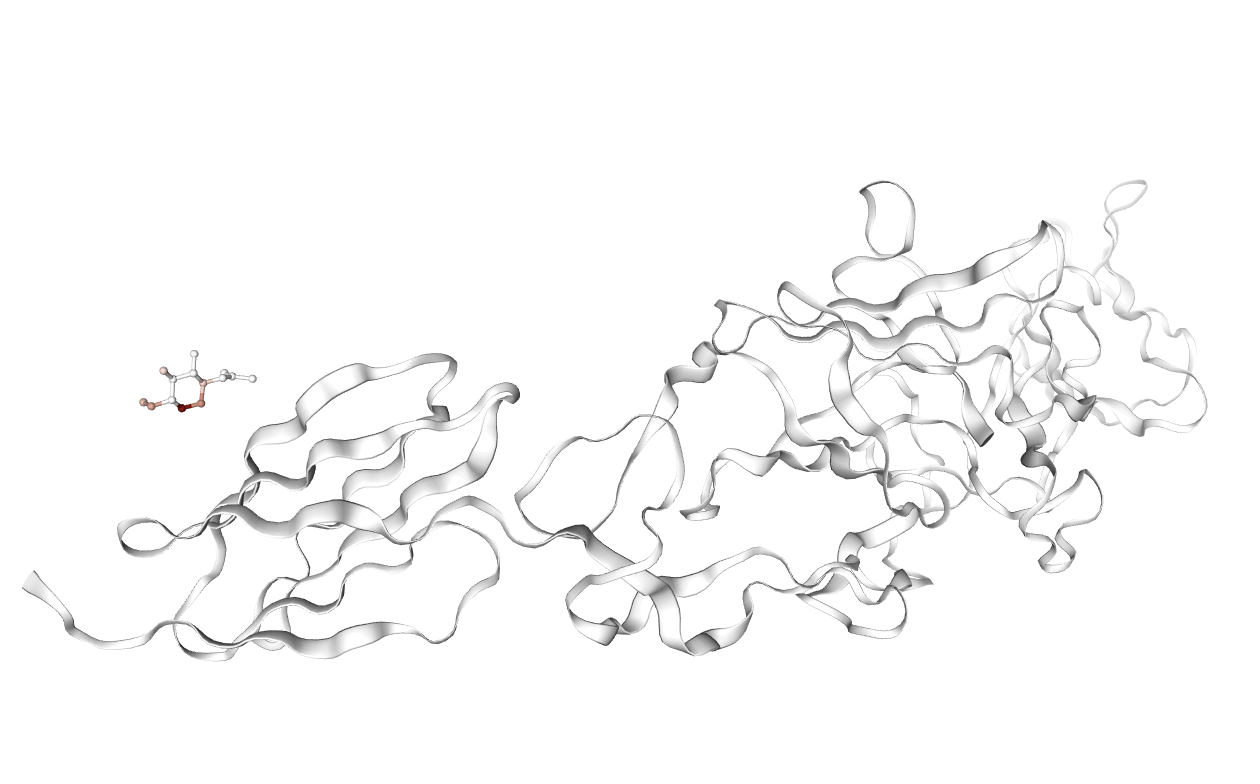
Figure 6: Representative examples of complexes where affinity-associated features are activated; highlighted molecules correspond to aberrant docking poses, underscoring potential failure cases for virtual screening.
Practical and Theoretical Implications
PairSAE advances mechanistic interpretability methodologies for protein structure foundation models by: (1) bridging information from both sequence and pair representations; (2) providing interpretable latent concepts directly aligned with annotated biophysical features; (3) supporting hypothesis generation for binding affinity prediction at high accuracy and sparsity levels.
Practically, this framework enables principled evaluation and debugging of large structure prediction models, accelerates functional screening, and suggests pathways for steerable protein design—particularly in scenarios where ligand annotation is sparse or absent. Theoretically, PairSAE highlights the utility of multilinear SVD compression and joint dictionary learning in disentangling polysemantic representations in bioinformatics models. The methodology is extensible to further recycling steps, additional layers, and automated interpretability pipelines.
Future Directions
The paper identifies open questions: scaling PairSAE to deeper layers and full model recycling, mapping ligand-activating features to precise molecular concepts (once annotation datasets improve), and integrating feature interventions for steerable protein or ligand design. Extension to multi-sequence alignments and incorporation into retrosynthetic pipelines offer prospects for future research.
Conclusion
PairSAE establishes a rigorous dictionary learning mechanism for mechanistic interpretability in pairformer-style protein models, unlocking token-level features predictive of both structural annotation and binding affinity. Its efficient architecture and robust methodology carry significant implications for virtual screening, annotation-driven model analysis, and future steerable molecular design paradigms.

