- The paper presents a machine-learned coarse-grained model that integrates temperature-dependent constraints to decompose the PMF into energetic and entropic components.
- It employs graph neural networks with temperature-dependent priors to accurately reproduce free energy landscapes and equilibrium state populations over multiple thermal regimes.
- The framework robustly predicts temperature-dependent observables, such as isochoric heat capacity, and enables rapid adaptation to new thermal conditions.
Thermodynamic Transferability in Machine-Learned Coarse-Grained Protein Models
Introduction
Machine-learned coarse-grained (MLCG) models have emerged as a powerful paradigm to simulate protein dynamics efficiently, outperforming traditional approaches by leveraging graph neural networks (GNNs) to capture many-body interactions. Despite advances, standard MLCG models remain constrained to single thermodynamic state points, lacking explicit temperature transferability, impeding their ability to accurately predict temperature-dependent observables such as heat capacity or free energy landscapes across thermal regimes. The paper "Temperature transferable Machine Learned Coarse Grained model for proteins" (2606.14111) addresses these deficiencies by proposing a thermodynamically informed MLCG framework that enforces a rigorous energetic-entropic decomposition of the potential of mean force (PMF). This architecture factors in exact thermodynamic relations, enabling physically consistent interpolation and extrapolation over temperature.
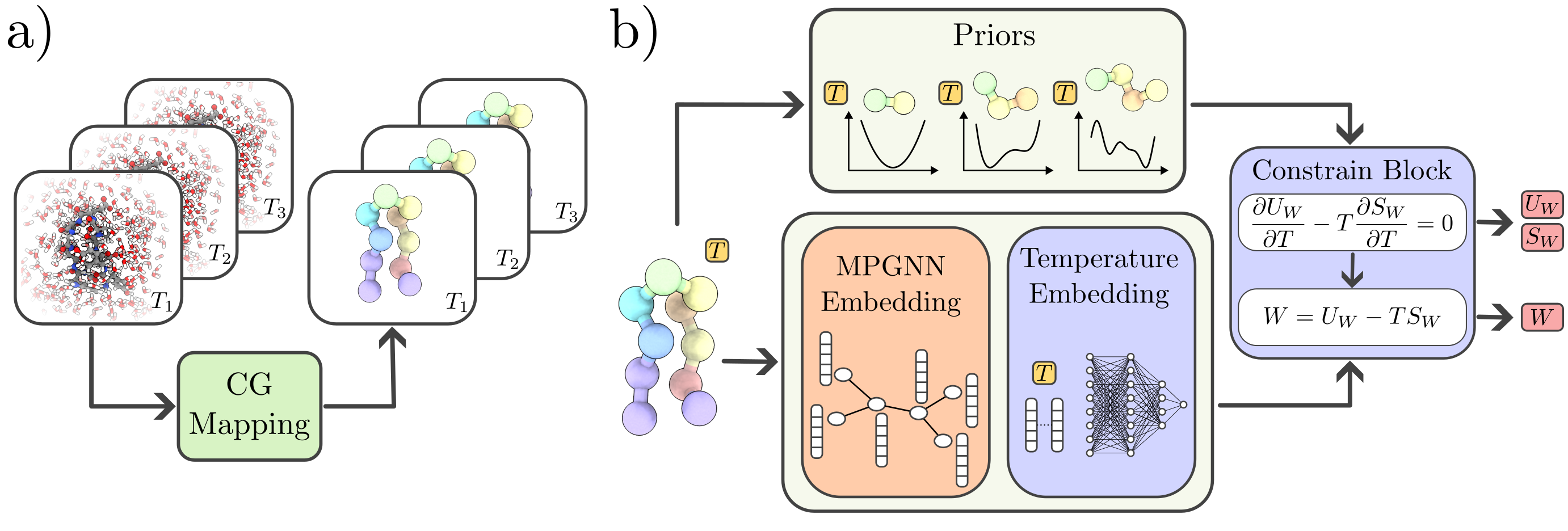
Figure 1: Schematic overview of the temperature-transferable MLCG model, illustrating multi-temperature dataset projection, architecture extensions (temperature-dependent priors, temperature embedding, thermodynamic constraint block).
Model Architecture and Thermodynamic Constraints
The foundation of the approach is a decomposition of the configuration-dependent PMF into energetic (UW) and entropic (SW) components:
W(R)=UW(R)−TSW(R).
The temperature dependency of the PMF, dominated by the entropic term, dictates thermodynamic transferability. The GNN-based MLCG model is constructed to produce not only approximations of the PMF via force matching, but also explicit energetic contributions using energy matching. The architecture includes temperature-dependent prior models for bonds, angles, and dihedrals, a temperature embedding layer, and a constraint mechanism that enforces the Maxwell-type thermodynamic relation:
∂T∂UW−T∂T∂SW=0.
Instead of penalizing violations via soft constraints during optimization, the constraint is enforced by design in the neural network output, yielding PMF predictions that remain physically consistent across temperature regimes.
Temperature-Dependent Priors and Dataset
To stabilize MD simulations and allow physical extrapolation, the model includes temperature-dependent prior terms. Priors are analytically derived or estimated via Boltzmann inversion for features such as bonded distances under Gaussian assumptions, yielding priors with linear or higher-order temperature dependence. Notably, spatially uniform temperature-dependent shifts are introduced, crucial for correcting energy baselines and enabling accurate prediction of thermodynamic observables without retraining.
The model is trained on an extensive dataset of atomistic molecular dynamics for the chignolin protein, spanning five temperatures (\SI{300}{\kelvin} to \SI{400}{\kelvin}) and totaling 250 μs of aggregated simulation. A Cα mapping integrates out side-chain atoms and solvent, and all relevant thermodynamic quantities are recorded every 2 ps.
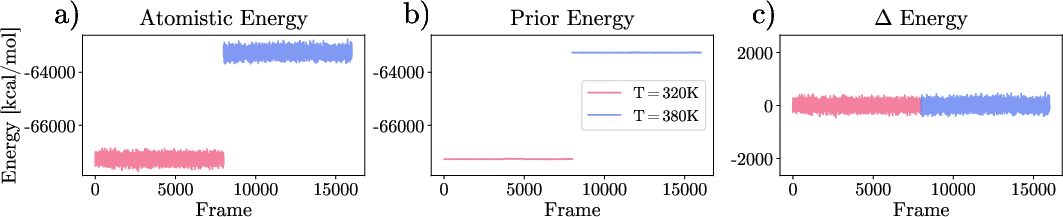
Figure 2: Prior effects in Delta-energy calculation, displaying atomistic reference, prior energy profiles, and resulting Delta-energies for two training temperatures.
Numerical Evaluation: Free Energy Landscapes and Thermodynamic Consistency
MD simulations performed with the trained temperature-transferable MLCG model and standard single-temperature baselines are evaluated in terms of their ability to reproduce atomistic free energy surfaces and equilibrium distributions across temperature. The free energy is projected along the first TICA component and clustered into folded, unfolded, and misfolded states. Single-temperature models fail to preserve correct populations away from their training conditions, e.g., over-stabilizing unfolded states at higher temperatures. In contrast, the temperature-dependent model accurately recovers atomistic balance between states across interpolation and extrapolation regimes.
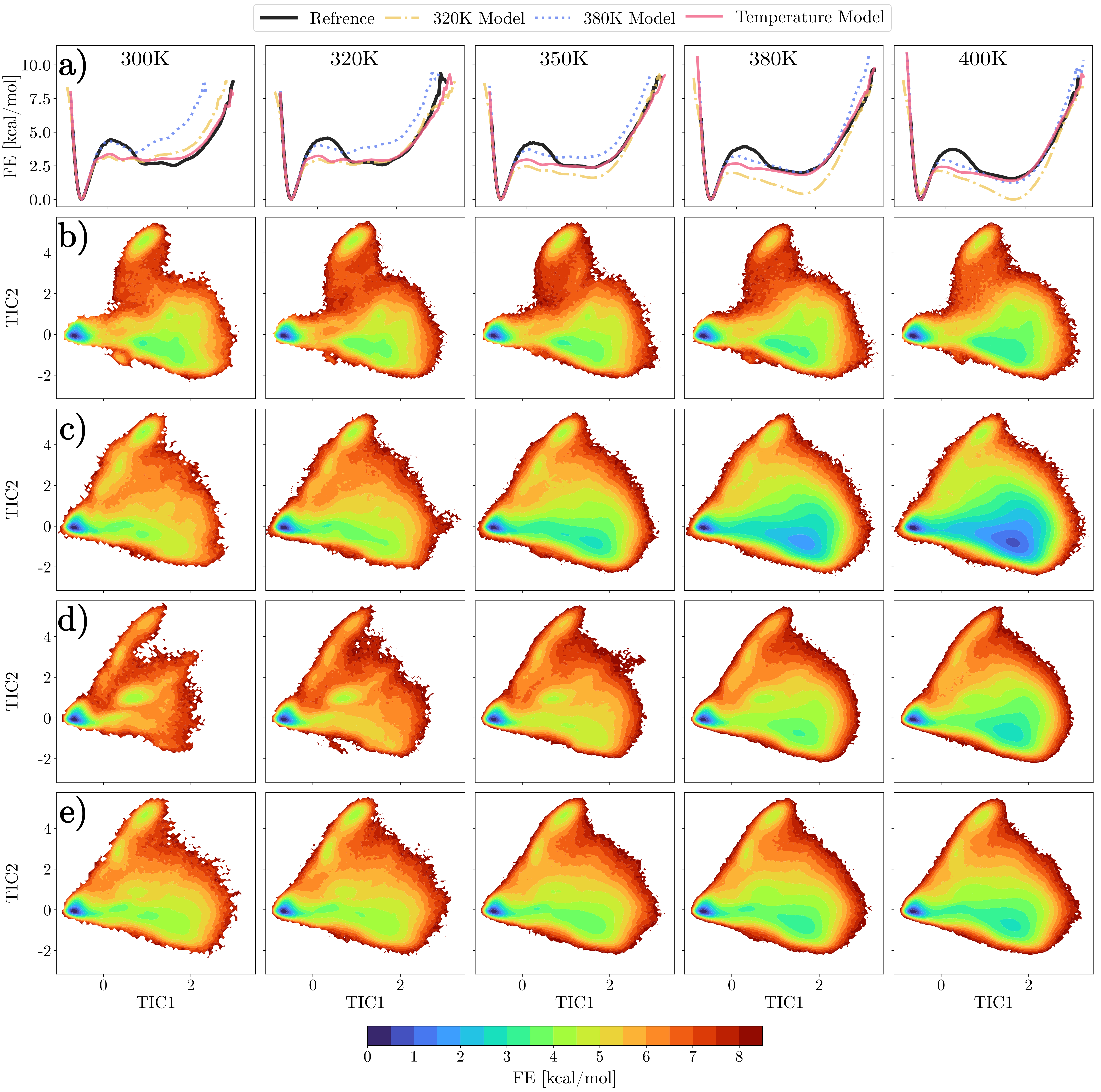
Figure 3: Free energy predictions in TICA space across different models and temperatures, highlighting preservation of folded/unfolded state balance only with temperature-dependent MLCG.
Errors in relative stability (ΔG) between folded/unfolded states are quantified. The temperature-dependent model shows excellent agreement with atomistic reference for ΔG across the investigated range, while single-temperature baselines exhibit strong deviations, including sign errors in extrapolation.
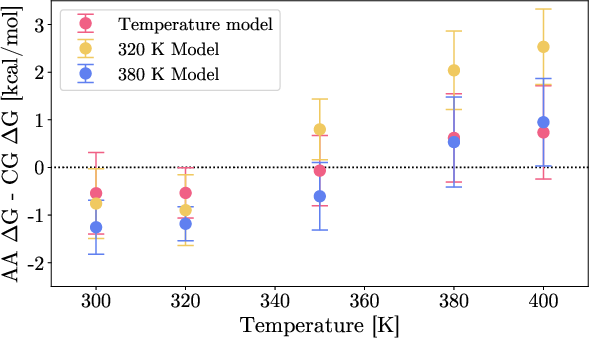
Figure 4: Difference in ΔG between atomistic reference and CG models across temperatures, showing consistency only in the temperature-dependent model.
Prediction of Thermodynamic Quantities
A critical advantage of the framework is its ability to predict temperature-dependent thermodynamic observables, such as isochoric heat capacity (cV), which standard CG models cannot provide. The mean internal energy, and thus cV, is computed from the energetically explicit component SW0. Linear shifts in prior energy yield inaccuracies outside training points; however, post-hoc correction via higher-order polynomial fitting to atomistic mean energies, using minimal additional data, recovers SW1 with high fidelity.
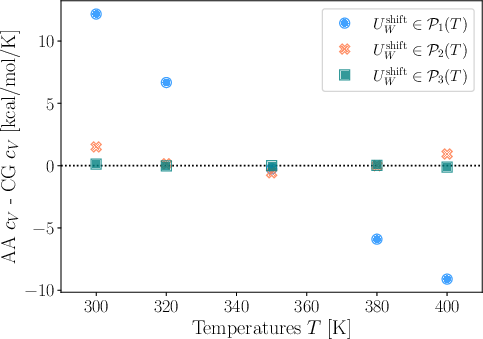
Figure 5: Absolute differences between atomistic and CG heat capacities, demonstrating accuracy only when higher-order temperature shifts are included in the CG model.
Polynomial corrections for global energetic shift decouple structural features learned by the GNN from global energetic baselines, facilitating rapid, accurate adjustment of thermodynamic observables without retraining.
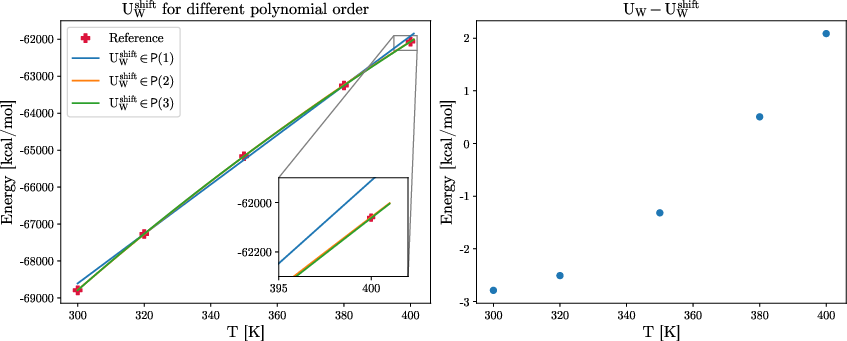
Figure 6: Contributions of different polynomial orders to SW2 and mean potential energy predicted by the GNN, excluding global temperature shift.
Implications and Future Directions
The presented model provides a robust pathway toward physically grounded, thermodynamically transferable MLCG simulations for proteins, combining rigorous energetic-entropic decomposition, constrained neural network architecture, and modular prior corrections. The enforcement of thermodynamic relations by construction, rather than via regularization, ensures validity outside training domains for both structural and thermodynamic predictions. The methodology addresses core issues of transferability and representability in CG modeling, and paves the way for new applications in protein engineering, biomolecular thermodynamics, and temperature-adaptive simulation protocols.
Practically, the architecture is compatible with data-efficient scalar corrections to energy priors, facilitating rapid adaptation to new temperature regimes. Theoretically, the explicit separation of energetic and entropic contributions enables fundamental studies of how renormalized degrees of freedom contribute to thermodynamic quantities, revealing that heat capacity is dominated by structure-independent energetic shifts while temperature-dependent balance between states requires explicit temperature dependence in the force field.
Future directions include extending the model to more complex proteins, including solvent effects, expanding beyond the canonical ensemble, and integrating uncertainty quantification for predictions in unexplored state points. The approach may be generalized to facilitate transferability with respect to other control parameters such as pressure or solvent conditions.
Conclusion
The temperature-transferable MLCG framework enables physically consistent, thermodynamically extrapolatable CG simulations of proteins by enforcing explicit energetic-entropic decomposition and thermodynamic constraints within a GNN architecture. The model accurately reproduces atomistic free energy surfaces, equilibrium state populations, and thermodynamic observables across a broad temperature range, with strong numerical agreement for heat capacity and relative stability. Modular energetic prior corrections further decouple structural learning from global energetics, allowing rapid adaptation to new conditions. The work addresses longstanding limitations in CG modeling, providing both practical and theoretical advances with implications for biomolecular simulation and thermodynamics.