- The paper establishes an extensible benchmark for rigorously evaluating deep learning models in single-cell multi-omics modality translation.
- It employs multi-faceted metrics, including clustering, regression, and distribution similarity, to reveal trade-offs between biological resolution and quantitative fidelity.
- Empirical findings highlight the impact of model architecture and feature quality, guiding future improvements in imputation and network inference for multi-omics data.
scTranslation: A Task-Oriented Benchmark for Single-Cell Multi-Omics Modality Translation
Introduction
Single-cell technologies have accelerated the characterization of regulatory programs through direct measurement of DNA accessibility (ATAC-seq), gene expression (RNA-seq), and protein abundance (Protein-seq) in individual cells, forming the empirical basis for regulatory network modeling (Figure 1). However, high costs, data sparsity, and protocol complexity limit routine experimental multiplexing, making computational cross-modality translation a critical approach for inferring regulatory or phenotypic layers from partially observed data. Despite rapid advances in deep learning–based translators, the absence of a systematic benchmark impedes robust algorithmic comparison. "scTranslation: A Comprehensive Benchmark for Single-Cell Multi-Omics Modality Translation" (2606.03906) establishes a unified and extensible evaluation framework, enabling stratified assessment of state-of-the-art translation models across diverse biological axes, metrics, and noise regimes.
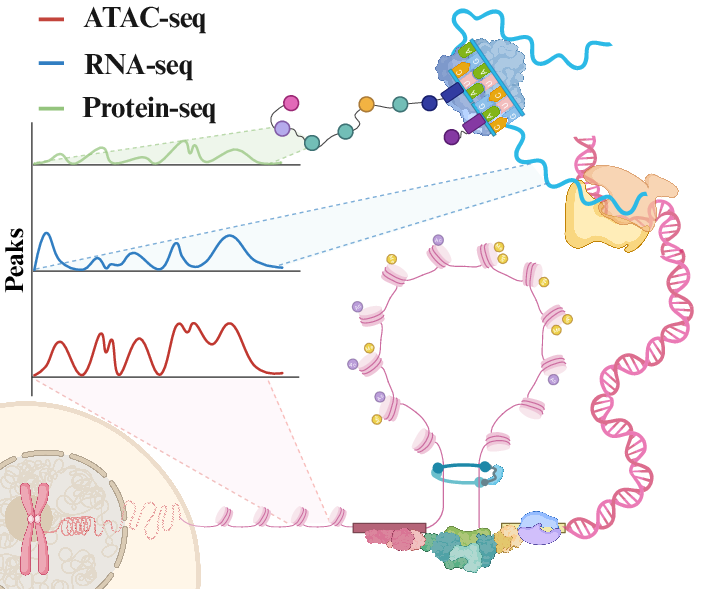
Figure 1: Schematic representation of the central dogma and the major single-cell modalities, highlighting ATAC-seq, RNA-seq, and Protein-seq.
Task Definition and Challenges
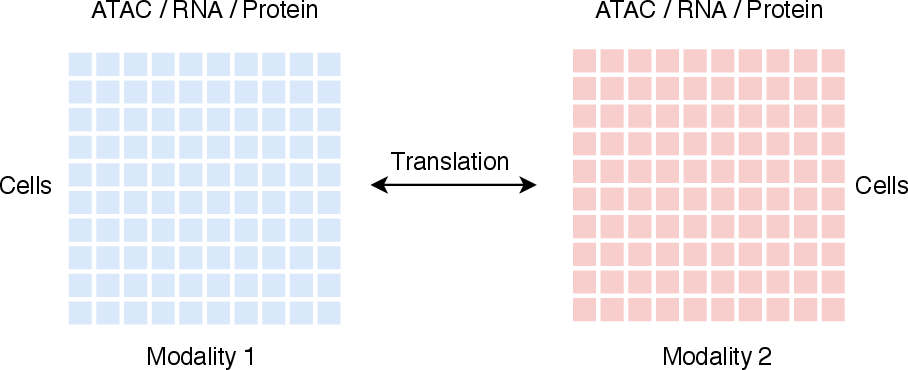
Cross-modality translation is formally posed as the mapping of a cell-by-feature matrix from source to target omics (e.g., RNA→ATAC, ATAC→RNA, RNA↔Protein) using paired measurements, seeking to preserve both biological specificity and quantitative fidelity (Figure 2). The dominant challenges addressed include:
Datasets and Benchmark Structure
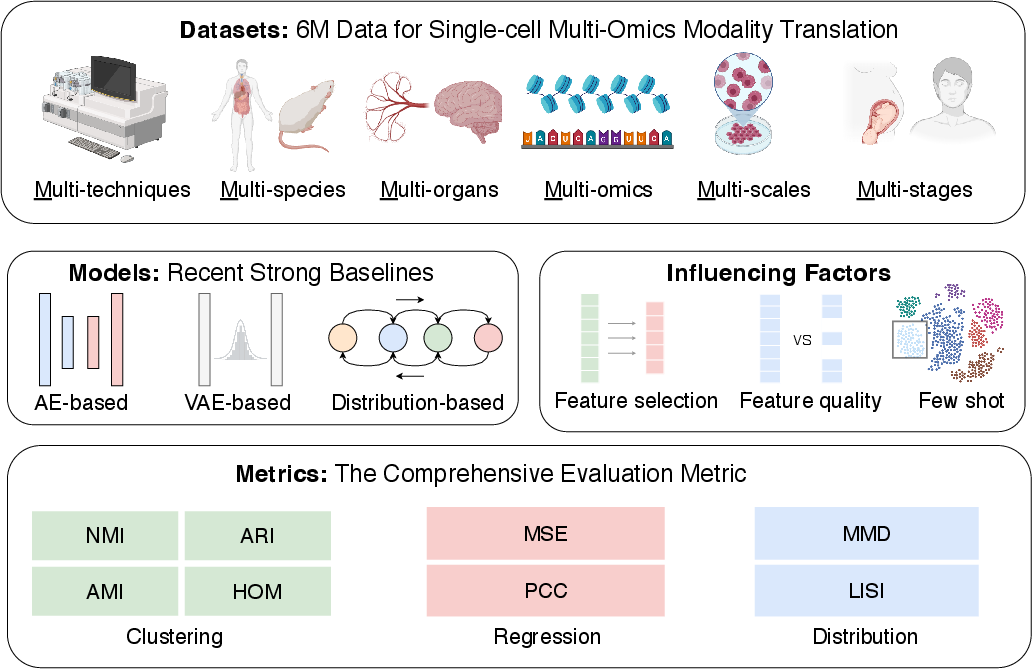
The authors curate eight public datasets covering a broad spectrum of organisms (mouse, human), organs (brain, PBMC, bone marrow), protocols (10x Multiome, SNARE-seq, sci-CAR, CITE-seq, scCAT-seq), and multi-omics layers (ATAC, RNA, Protein). The selection follows the "6M" criteria (technique, species, organ, modality, scale, developmental stage) to enable rigorous cross-context evaluation.
scTranslation organizes benchmarking in a modular pipeline (Figure 3):
Model Landscape and Methodological Spectrum
Baseline models are classified according to underlying generative approaches:
- Autoencoders (AE): BABEL, scPair rely on encoder-decoder architectures with joint latent spaces and adversarial alignment to support bidirectional translation.
- Variational Autoencoders (VAE): JAMIE and scButterfly employ probabilistic latent representations and explicit latent-space alignment, with JAMIE supporting missing modality imputation and scButterfly leveraging dual-aligned, semi-supervised VAE frameworks.
- Distribution-based Models: multiDGD (Gaussian mixture modeling), scDiffusion-X (diffusion process + cross-attention with optional label conditioning). These prioritize global data distribution matching.
Comprehensive Metrics for Biological and Quantitative Fidelity
The benchmark implements three metric groups:
- Clustering: NMI, ARI, AMI, HOM—probes conservation of cell identity and biological structure post-translation.
- Regression: PCC, MSE—quantifies the fidelity of feature-level prediction and reconstructive accuracy.
- Distribution: MMD, LISI—assesses global and local distributional concordance, critical for downstream integration and batch correction.
Empirical Results and Influencing Factors
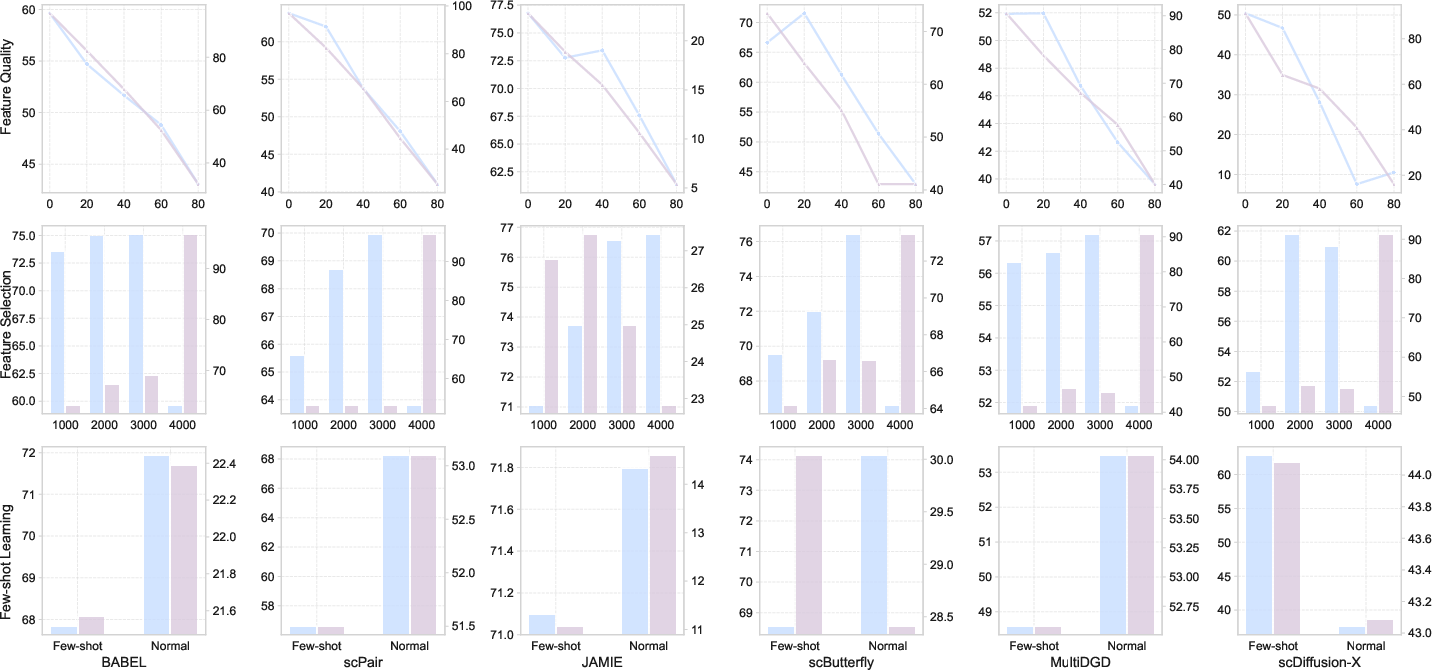
Supervised experiments demonstrate several key findings (Figure 4):
Practical and Theoretical Implications
scTranslation exposes intrinsic trade-offs between biological resolution and quantitative or global concordance, substantiating the claim that current approaches have not converged on a uniformly optimal regime. The results suggest the importance of robust architecture selection in routine biological applications such as multi-omic imputation, legacy dataset augmentation, and high-confidence network inference. Notably, translation performance is highly contingent on both input feature quality and the task’s data regime, indicating the need for adaptive, context-aware models in the future.
In the context of foundation models for biomedicine and generative AI, these findings underscore the necessity for large-scale, context-diverse multi-omic integration during pre-training and for evaluating model robustness under experimental noise, feature corruption, and data starvation. The limitations of current metrics (e.g., MSE overemphasizing amplitude, LISI missing rare population fidelity) call for future research into biologically grounded, composite evaluation frameworks.
Conclusion
scTranslation delivers a rigorous and extensible benchmarking system for single-cell multi-omics modality translation. By integrating task diversity, comprehensive metrics, and systematic robustness studies, it provides the necessary infrastructure for reproducible comparison, illuminates decisive factors affecting model performance, and outlines the major gaps remaining in neural-based cross-modality inference. The benchmark is well-positioned to catalyze the development of robust, generalizable, and biologically interpretable translation models in high-dimensional omic data analysis.
Reference:
"scTranslation: A Comprehensive Benchmark for Single-Cell Multi-Omics Modality Translation" (2606.03906).