- The paper demonstrates forward mapping using ALIGNN, achieving a mean MAE of 0.032 with 88% of test predictions below MAE 0.05.
- The inverse module employs a QLoRA-fine-tuned LLM, preserving chemical formulae in 86.8% of cases for practical motif recovery.
- RamanGPT’s unified round-trip workflow integrates spectrum retrieval, generative structure prediction, and real-time validation for advanced materials screening.
Framework Overview
RamanGPT operationalizes end-to-end computational workflows for crystalline Raman spectroscopy by integrating three major modules: (1) cosine-similarity retrieval against the Computational Raman Database (CRD, 5,099 DFPT spectra), (2) generative structure prediction via QLoRA-fine-tuned LLMs (AtomGPT), and (3) forward Raman spectrum prediction with an Atomistic Line Graph Neural Network (ALIGNN). These modules facilitate both structure-to-spectrum and spectrum-to-structure mappings, and are deployed through a public web interface supporting inverse→relax→forward round-trip validation.
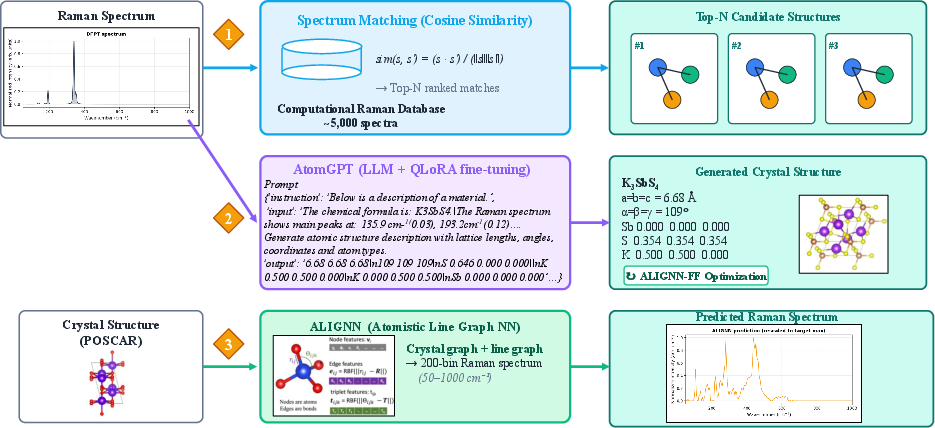
Figure 1: Overview of the RamanGPT framework, comprising spectrum matching, generative structure prediction, and forward spectral prediction modules.
The forward module leverages ALIGNN, which enriches message passing with both bond and bond-angle information, enabling accurate representation of crystal vibrational properties. The model is trained on 200-bin discretized Raman spectra from the CRD (50–1000 cm−1). Across 509 held-out test materials, the mean MAE reaches 0.032 (median: 0.029); 88% fall below MAE=0.05 and 99.4% below MAE=0.10. Model quality is better quantified through cosine similarity due to inherent spectral sparsity: 42.5% of predictions surpass cosine similarity ≥ 0.354, marking the threshold for qualitatively reasonable spectral matching. Notably, only 0.2% reach near-perfect matches.
Visual analysis reveals that peak broadening and smoothing are pronounced for complex spectra, while light-element compounds with high-frequency vibrational modes are less faithfully reproduced due to training-band truncation. Prediction fidelity declines primarily in regions with high Raman activity (100–400 cm−1) and improves above 600 cm−1 as spectra become increasingly sparse.
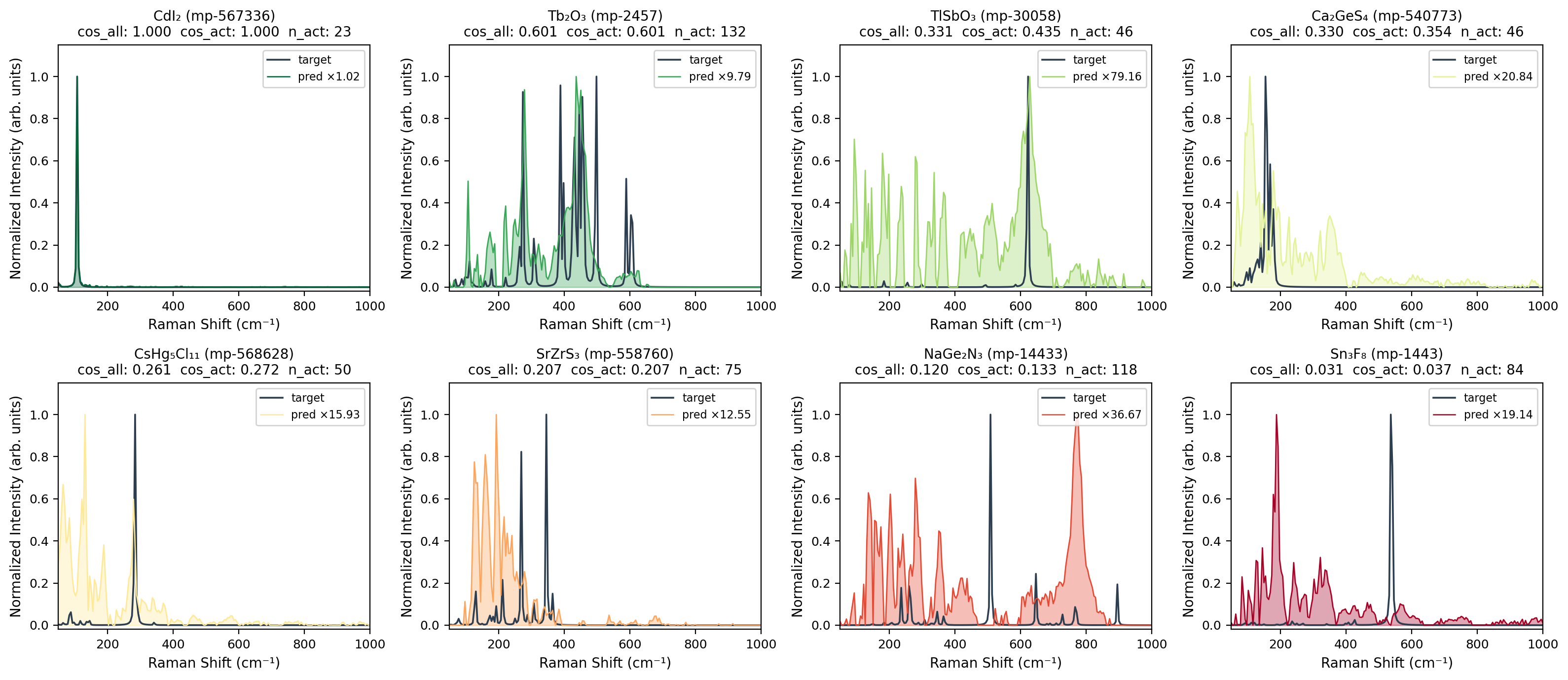
Figure 2: Forward Raman spectrum predictions (ALIGNN) across varying similarity regimes, demonstrating qualitative to near-quantitative reproduction of DFPT-computed spectra.
Element-wise cosine similarity analysis highlights superior model performance for lanthanides, chalcogens, and late transition metals, attributed to their localized, low-frequency modes. Performance degrades for light elements and actinides, often due to broader, dense multi-peak spectra or features outside the prediction window. Some periodic trends are attributable to dataset imbalance.
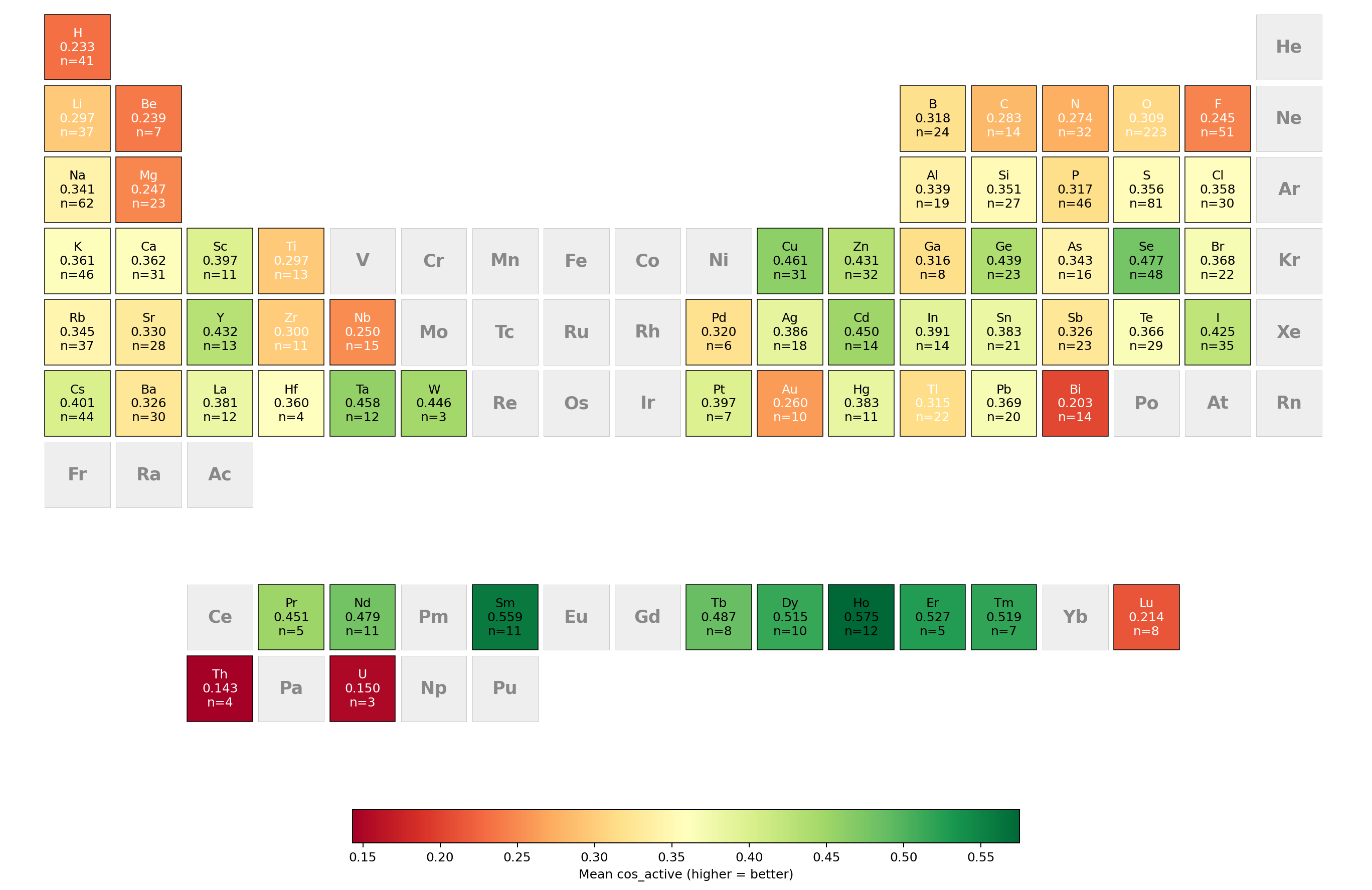
Figure 3: Periodic table heatmap of mean active-region cosine similarity (cos_active) for ALIGNN predictions per element, illustrating chemical-space performance variability.
The model is validated out-of-distribution on 1T VSe2 (metallic, absent from training), indicating qualitative alignment: predicted modes are shifted but retain relative intensities consistent with experiment, underscoring extensibility to novel materials.
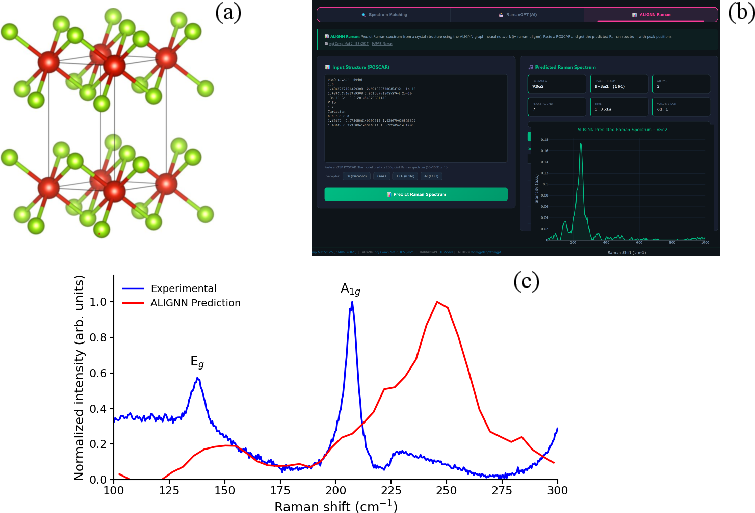
Figure 4: Comparison of ALIGNN-predicted and experimental Raman spectra for VSe2, validating model generalization.
The inverse module employs Mistral-7B-Instruct fine-tuned via QLoRA, ingesting Raman spectra paired with explicit chemical formula prompts (Alpaca-style). On 508 test materials, lattice parameter MAEs are 1.14–2.16~\AA{} (factor 2–7 higher than DiffractGPT's PXRD-based metrics), reflecting the more indirect mapping between vibrational features and atomic positions. Lattice-angle prediction is less precise (MAE 17–21∘), attributable to bias from mostly orthogonal CRD entries and limited spectral sensitivity to angular geometry.
Reduced formula is correctly preserved in 86.8% of cases (mean element accuracy: 76.3%). Fractional-coordinate RMSE is ~0.265, corresponding to 1.5–3~\AA{} of positional error, sufficient for motif identification but not crystallographic refinement. Space-group match rate is 18.9%, demonstrating sensitivity to small perturbations. Element composition and cell density distributions closely reproduce target values.
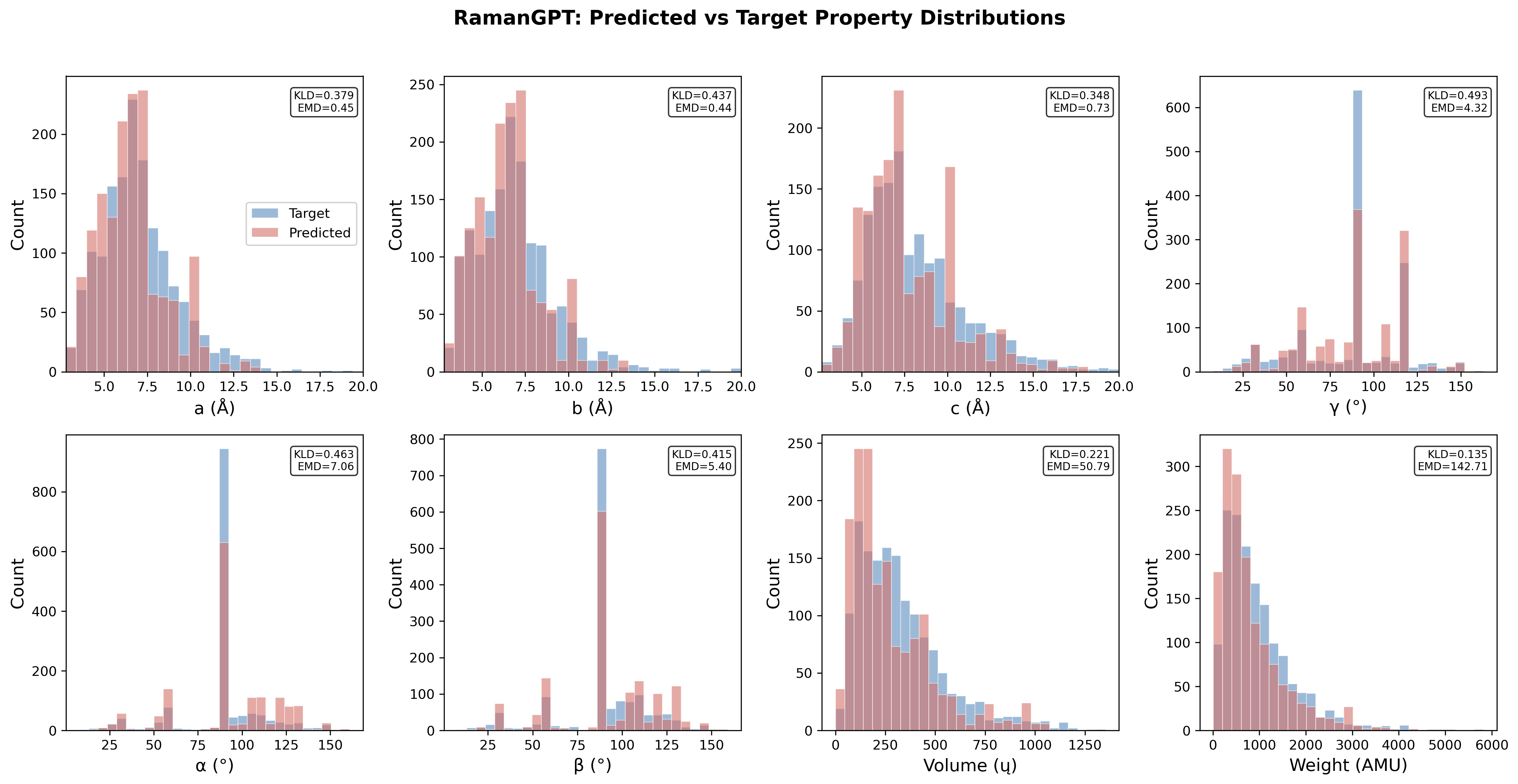
Figure 5: Distributions of predicted vs. target structural quantities for the inverse model, annotating KLD and EMD for agreement quantification.
The generative approach outperforms retrieval-only matching (formula: 41%, space group: 9%) and provides extrapolation capability beyond database entries. Prompt conditioning with formula results in canonicalization acccuracy rather than spectrum-driven element inference; formula-free element-list and ablation studies are deferred.
Practical and Theoretical Implications
RamanGPT establishes computational feasibility for high-throughput Raman spectrum prediction and structure recovery from vibrational data in crystalline systems. The framework provides near-real-time spectrum generation (ALIGNN) and generative structure suggestions (LLM)—significant for domains with incomplete reference databases (e.g., novel compounds, defected solids, or disordered phases).
Limitations include restriction to band-gap-above-0.5~eV, dynamically stable inorganic crystals. Expansion to metals and disordered systems requires resonance-aware descriptors or data augmentation. Windowing to higher frequencies necessitates increased model context and retraining. Angle bias in inverse predictions motivates auxiliary conditioning, and further experimental validation against broad-spectrum datasets remains critical for robustness.
Theoretical implications stem from demonstrated invertibility: although LLMs cannot reconstruct atomic detail at PXRD-level accuracy from Raman spectra alone, explicit conditioning yields practical approximations for motif identification and downstream relaxation/refinement pipelines. The unified deployment, including round-trip inverse→relax→forward validation, informs confidence metrics for practical use in materials screening and synthesis guidance.
Future Directions in AI for Materials Spectroscopy
The RamanGPT pipeline is extensible to infrared, terahertz, and inelastic neutron spectroscopy by database modification. Prompt templates may be broadened to include PXRD or microscopy input for multi-modal structure inference. Element-list and formula-free ablation studies, improved angle prediction via crystal-system conditioning, and broader experimental validation against laboratory datasets will further assess and enhance generative accuracy.
Conclusion
RamanGPT advances integration of deep graph networks and LLMs for both spectrum prediction and generative structure recovery in crystalline Raman spectroscopy—the dominant vibrational probe in materials science. Forward module performance achieves qualitative to semi-quantitative spectral reproduction for a substantial fraction of materials. Inverse generative modeling demonstrates practical motif recovery, albeit with limited precision compared to diffraction-based analogues. The framework's unified, open deployment enables advanced retrieval, prediction, and generative inversion workflows, and provides a foundation for further extension to new spectroscopic regimes and experimental validation (2606.03764).

