- The paper introduces CSP-MACE-Å, a ML interatomic potential that decomposes total energy into intra- and intermolecular components to achieve DFT-equivalent accuracy.
- It leverages MACE-POLAR and a delta correction model, demonstrating near-parity with DFT on AstraZeneca and blind-test crystal datasets.
- The method accurately predicts temperature-dependent polymorphic stability, significantly accelerating CSP workflows and supporting advanced solid-form risk analysis.
DFT-Level Accuracy in Crystal Structure Prediction: CSP-MACE-Å Machine Learning Potential
Model Architecture and Methodological Innovations
The manuscript presents CSP-MACE-Å, a machine learning interatomic potential (MLIP) targeting DFT-level accuracy for crystal structure prediction (CSP) workflows. The model employs a decomposition of total energy into intramolecular and intermolecular components, allocating dedicated architecture and training protocols for each interaction type. The intramolecular term is modeled using MACE-POLAR, trained on the OMol25 dataset (100M ωB97M-V/def2-TZVPD DFT calculations). The intermolecular component integrates three terms: a MACE-POLAR intermolecular contribution, a fixed-parameter XDM-form dispersion correction, and a delta model trained to correct residuals to B86bPBE-XDM DFT intermolecular energies using 50,000 crystal structure calculations.
This approach directly addresses two longstanding limitations of MLIPs for CSP: explicit modeling of long-range interactions (dispersion and electrostatics) and accurate parameterization for subtle intermolecular energies in crystals. The delta model training on isolated intermolecular signals overcomes gradient dilution from dominant intramolecular contributions—a problem in standard MLIP protocols for crystals.
Evaluation on AstraZeneca and CSP Blind Test Compounds
CSP-MACE-Å was benchmarked against dispersion-corrected DFT (PBE-NP and B86bPBE-XDM) and MLIP baselines (MACE-POLAR-1 and UMA-OMC) on two curated sets:
- AZ Set: 19 compound structures from AstraZeneca CSP studies, spanning large candidate pools and including salts.
- Blind Test Set: 28 molecule crystals from the seven CSP blind tests, comprising cocrystals, salts, and experimentally challenging instances.
CSP-MACE-Å achieved parity or near-parity with DFT on both datasets. For the AZ set, energy-based ranking consistently identified experimental matches within the top 10, and a ΔE within 2 kJ/mol. Free energy ranking (harmonic approximation at 300 K) improved accuracy further, outperforming all other MLIPs and matching or exceeding DFT metrics.
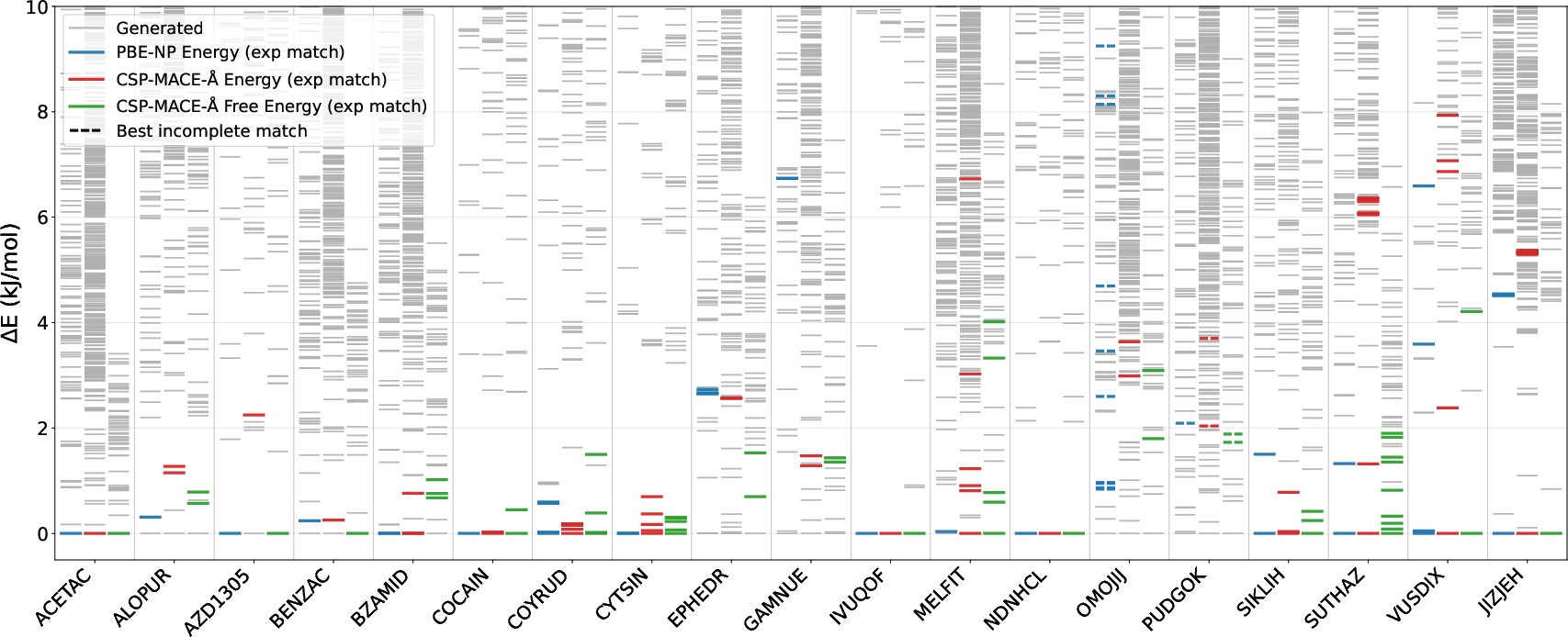
Figure 1: AZ evaluation set relative energy landscape comparing PBE-NP DFT, CSP-MACE-Å, and harmonic free energy rankings for each compound.
In the blind-test regime, CSP-MACE-Å performed similarly: while energy ranking lagged marginally behind B86bPBE-XDM, harmonic free energy reranking surpassed DFT rankings, a claim substantiated across all 28 test cases.
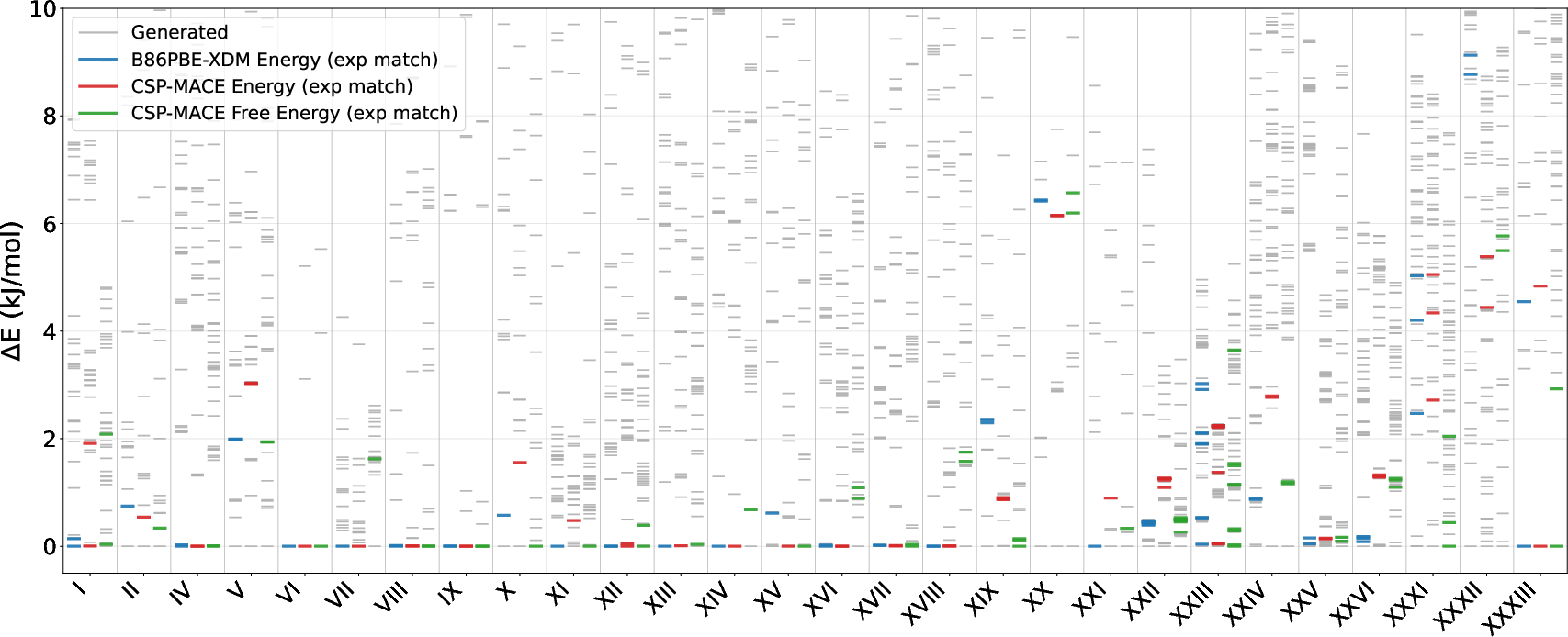
Figure 2: Blind test evaluation set relative energy landscape showing B86bPBE-XDM DFT, CSP-MACE-Å, and CSP-MACE-Å harmonic free energy rankings for each compound.
These numerical results demonstrate that CSP-MACE-Å achieves DFT-equivalent accuracy across both energy and free energy rankings with an inference speed advantage of several orders of magnitude.
Addressing Intramolecular and Intermolecular Modeling Challenges: The ROY Case Study
ROY (Red Orange Yellow) is a hallmark test for CSP, possessing 14 known polymorphs and exposing DFT-D delocalization errors. Previous studies show standard DFT-D methods erroneously rank Form Y—experimentally most stable—several kJ/mol above other forms. CSP-MACE-Å, leveraging the high-level ωB97M-V intramolecular model and B86bPBE-XDM intermolecular matching, does not suffer from this artifact. Form Y is ranked within 0.5 kJ/mol of the minimum, aligning with experimental stability.
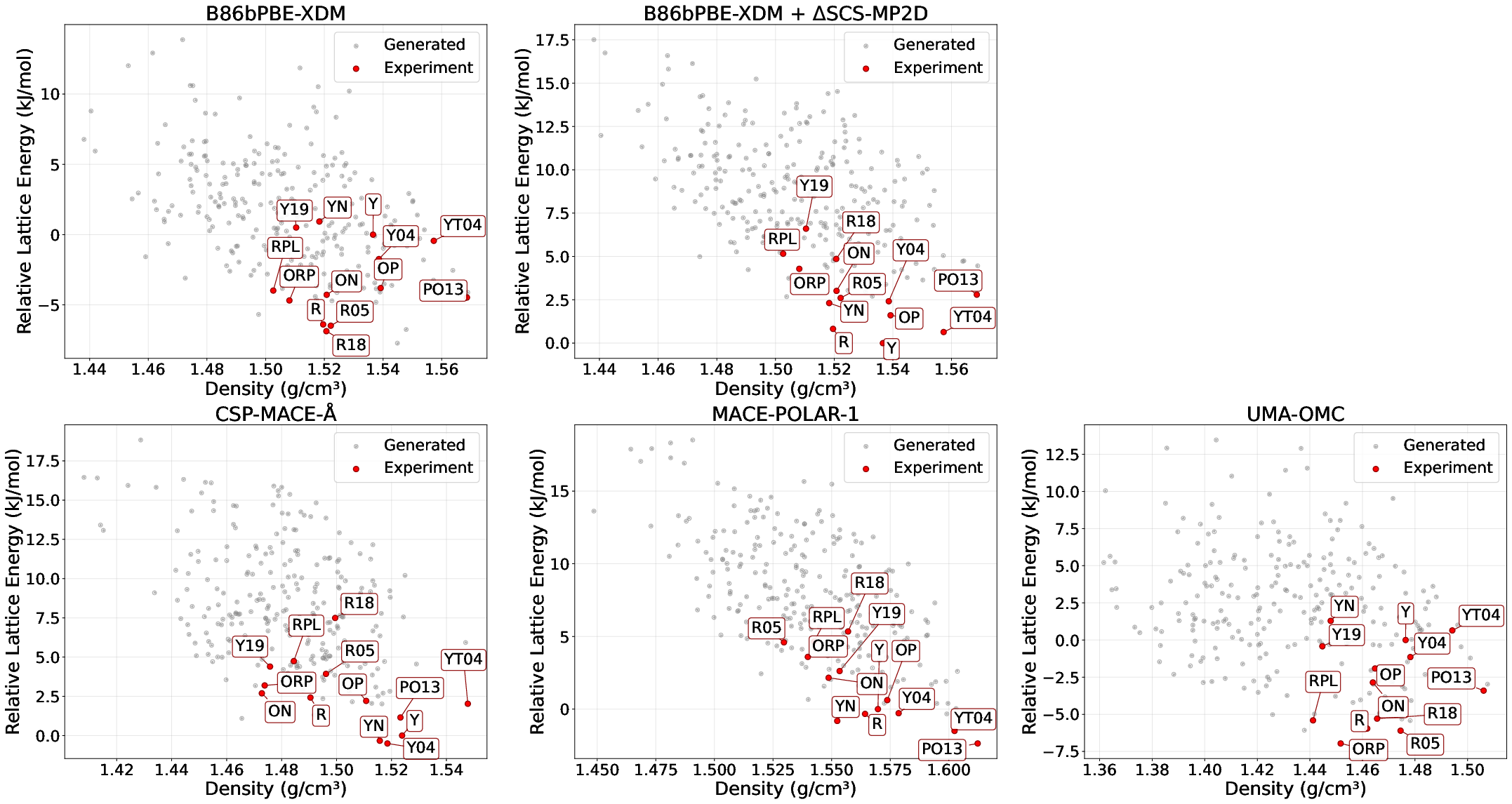
Figure 3: ROY energy landscape: Lattice energies (relative to Form Y) illustrate CSP-MACE-Å's resolution of DFT-D ranking failures.
UMA-OMC and B86bPBE-XDM fail to remedy the intramolecular error, confirming CSP-MACE-Å's superior architectural partitioning for accuracy-critical systems.
Thermodynamic Stability Predictions Across Polymorphic Landscapes
The model's extension to temperature-dependent polymorph stability was validated on five compounds spanning diverse pharmaceutical relevance (Sulfathiazole, Mexiletine hydrochloride, AZD1305, a Triazolo-pyrimidine, and AZD5462). For each system, CSP-MACE-Å computed harmonic free energies (0–600 K), tracking experimentally validated transitions and stability regimes.
For Sulfathiazole, CSP-MACE-Å reproduced the broad trend of stability shifts with temperature, though failed to resolve the exact polymorph ordering at extremes, paralleling inaccuracies of prior DFT protocols.
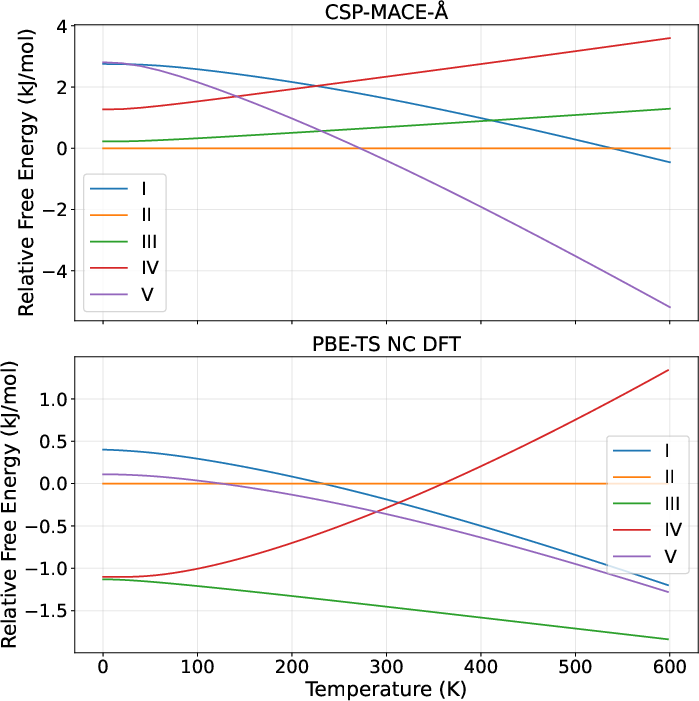
Figure 4: Sulfathiazole thermodynamic stability predictions with CSP-MACE-Å and PBE-TS NC DFT vs experimental temperature-dependent ordering.
Mexiletine hydrochloride, AZD1305, Triazolo-pyrimidine, and AZD5462 yielded predictions that closely matched experimental monotropic/enantiotropic behaviors, capturing the key transitions (though with modest range deviations in temperature intervals for certain forms).
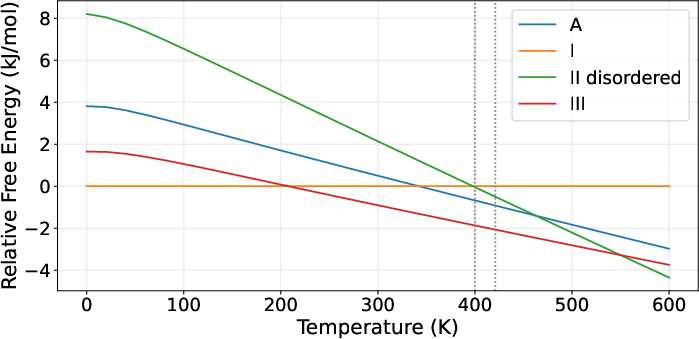
Figure 5: Mexiletine hydrochloride predictions: Relative free energy vs temperature, showing approximated experimental transitions.
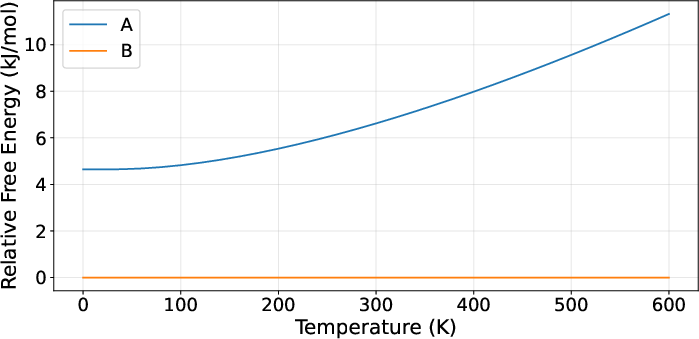
Figure 6: AZD1305 predictions: Monotropic relationship confirmed, though relative stabilities not fully convergent at high temperature.
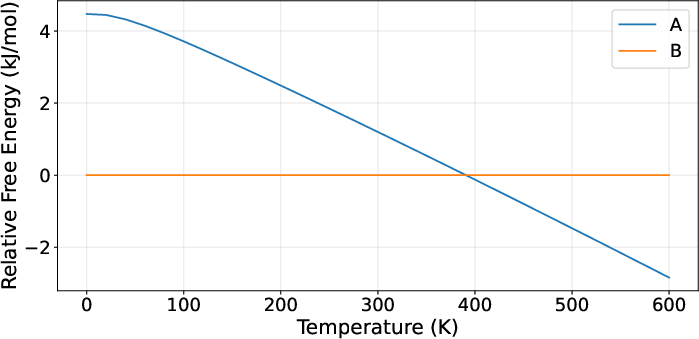
Figure 7: Triazolo-pyrimidine: Accurate stability reversal between Forms A and B with temperature.
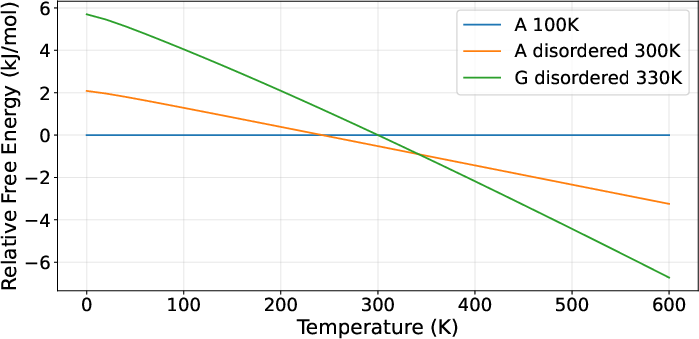
Figure 8: AZD5462: Stability curves for ordered and disordered forms accurately capturing experimental regimes.
Comparative RMSD and Structural Matching
Structural overlays and RMSD analyses confirm high fidelity matches between CSP-MACE-Å relaxed structures and experimental reference crystals, further consolidating the claim of DFT-level geometric accuracy.
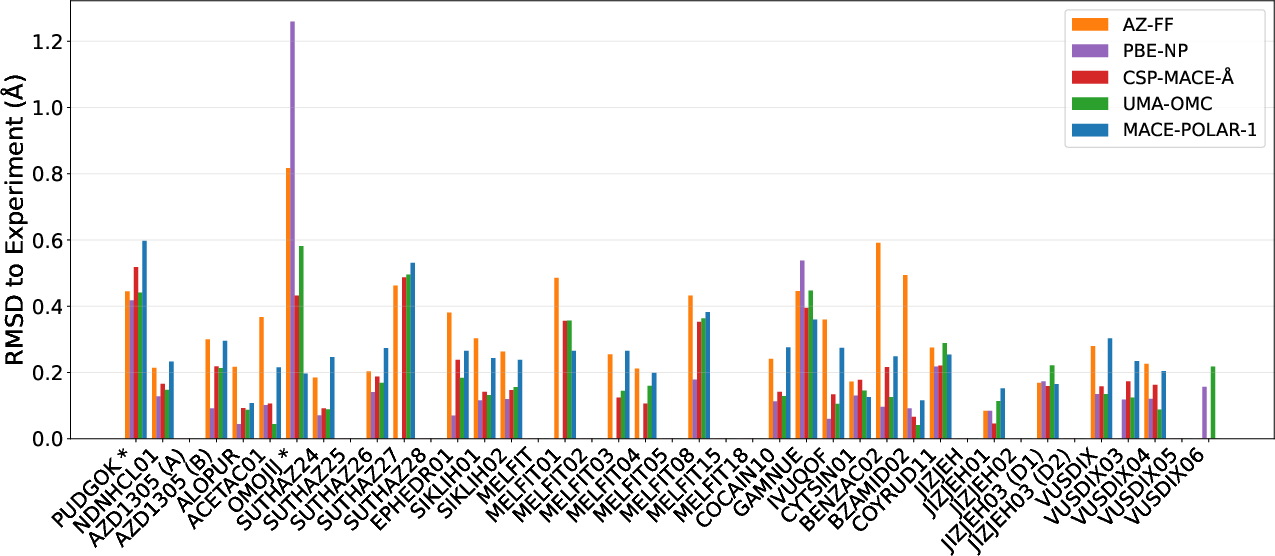
Figure 9: RMSD scores for relaxed structures matched to experimental forms across methods.
Implications, Limitations, and Future Directions
The results establish CSP-MACE-Å as the first MLIP capable of effectively replacing DFT in CSP workflows, resolving both intramolecular and intermolecular modeling pathologies that previously hindered MLIPs. The practical implication is a profound acceleration of CSP, enabling both richer candidate structure evaluation pools and feasible free energy ranking stages previously excluded by DFT cost. The model's ability to reproduce temperature-dependent polymorphic behavior also opens avenues for more comprehensive solid-form risk assessment and early-stage drug development integration.
From a theoretical standpoint, CSP-MACE-Å demonstrates that partitioned energy modeling and targeted delta learning are essential architectural innovations for MLIPs in crystalline molecular systems. The inclusion of explicit dispersion correction and intermolecular residual learning optimally addresses the complexity of crystal energetics.
Future research directions include re-engineering CSP workflows to exploit the speed and scalability of CSP-MACE-Å, such as integrating ML-based ranking earlier in structure generation stages and exploring coupling with property prediction modules (e.g., solubility). The architectural paradigm could generalize to other solid-state physics applications requiring accurate balance between intra- and inter-entity interactions.
Conclusion
The study rigorously evaluates CSP-MACE-Å as a machine-learned surrogate for DFT in crystal structure prediction. By decomposing energy components and explicitly modeling long-range interactions, the model achieves DFT-analogous accuracy for both energy and free energy rankings across established pharmaceutical benchmarks and blind test sets. CSP-MACE-Å resolves historic intramolecular modeling failures, as evidenced in ROY, and captures temperature-dependent stability trends. The computational efficiency and accuracy attained have substantial practical and theoretical implications, marking a substantial advance in ML-based CSP and solid-form risk mitigation for the pharmaceutical domain (2605.28905).

