OXtal: An All-Atom Diffusion Model for Organic Crystal Structure Prediction
Abstract: Accurately predicting experimentally-realizable 3D molecular crystal structures from their 2D chemical graphs is a long-standing open challenge in computational chemistry called crystal structure prediction (CSP). Efficiently solving this problem has implications ranging from pharmaceuticals to organic semiconductors, as crystal packing directly governs the physical and chemical properties of organic solids. In this paper, we introduce OXtal, a large-scale 100M parameter all-atom diffusion model that directly learns the conditional joint distribution over intramolecular conformations and periodic packing. To efficiently scale OXtal, we abandon explicit equivariant architectures imposing inductive bias arising from crystal symmetries in favor of data augmentation strategies. We further propose a novel crystallization-inspired lattice-free training scheme, Stoichiometric Stochastic Shell Sampling ($S4$), that efficiently captures long-range interactions while sidestepping explicit lattice parametrization -- thus enabling more scalable architectural choices at all-atom resolution. By leveraging a large dataset of 600K experimentally validated crystal structures (including rigid and flexible molecules, co-crystals, and solvates), OXtal achieves orders-of-magnitude improvements over prior ab initio machine learning CSP methods, while remaining orders of magnitude cheaper than traditional quantum-chemical approaches. Specifically, OXtal recovers experimental structures with conformer $\text{RMSD}_1<0.5$ Å and attains over 80\% packing similarity rate, demonstrating its ability to model both thermodynamic and kinetic regularities of molecular crystallization.



Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper introduces OXtal, a computer model that predicts how small organic molecules arrange themselves in 3D when they form crystals. It starts from a simple 2D drawing of a molecule (its “chemical graph”) and tries to guess the realistic 3D crystal structure you could observe in the lab. This matters because the way molecules pack together in a solid changes how that material behaves—important for medicines, electronics, and more.
What are the big questions the paper tries to answer?
The researchers aim to answer:
- Given only a 2D description of a molecule, can we quickly and accurately predict the 3D crystal structure it forms?
- Can a data-driven model learn the rules of crystal packing directly from many examples, instead of running very slow physics simulations?
- Can we model both the shape each molecule adopts inside a crystal and how multiple molecules line up and repeat in space?
How does OXtal work?
A quick primer on crystals and packing
Imagine building a repeating pattern with LEGO bricks on a baseplate. In a crystal, the “bricks” are molecules, and the baseplate is the invisible grid that defines how the pattern repeats in 3D (the “lattice”). Many organic crystals have lots of atoms per repeating block (the “unit cell”) and weak, long-range interactions between molecules—like bricks that don’t snap firmly but still settle into neat repeating patterns.
Two main challenges make this hard:
- Each molecule can bend and twist (its “conformation”), and this shape is influenced by how it’s packed with neighbors.
- The repeating pattern can be large and complex, and many different arrangements can be “good enough,” making the search space huge.
Diffusion models, explained simply
OXtal uses a diffusion model, a type of AI that learns to turn “random noise” into a clean, realistic structure step by step. Think of it like un-blurring a photo: the model practices removing noise so it can reconstruct the original picture. During training, it adds noise to real crystal structures and learns to reverse the process. During prediction, it starts from noise and “denoises” into a plausible crystal arrangement.
Training with “shells” (S4)
A key idea in this paper is Stoichiometric Stochastic Shell Sampling (S4). Here’s the intuition:
- Crystals grow from small clusters outwards, like how ice crystals form layer by layer.
- Instead of forcing the model to learn the entire huge repeating lattice, S4 crops local neighborhoods around a central molecule in expanding “shells” (layers of nearby molecules at increasing distances).
- These shells preserve the ratio of different molecule types (the “stoichiometry”) and expose the model to the specific local contacts that eventually create long-range repeating patterns.
- This makes training more scalable and helps the model learn the right local interactions without juggling fragile global lattice parameters.
Analogy: If you want to understand a city’s layout, you can learn a neighborhood at a time—side streets, parks, and shops—and still get a sense of the bigger pattern later.
The model pieces in plain terms
OXtal combines:
- An atom encoder: it represents each atom’s type and properties (like charge) and starts from a reasonable 3D guess for the molecule’s shape.
- A “Pairformer” trunk: a neural network that lets atoms “talk” to each other, sharing information about both single atoms and pairs of atoms. It’s inspired by models used to predict protein structures.
- A diffusion transformer: the main engine that turns noisy inputs into clean 3D positions for all atoms, eventually producing a plausible crystal crop.
Instead of hard-coding symmetry rules (like always rotating something exactly the same way), they use data augmentation (showing the model rotated and shifted versions of the same structures) so it learns symmetry naturally, which scales better to large problems.
What did they find?
The team trained OXtal on about 600,000 real, experimentally confirmed organic crystal structures, covering rigid molecules, flexible molecules, co-crystals (made of more than one molecule), and solvates.
Key results:
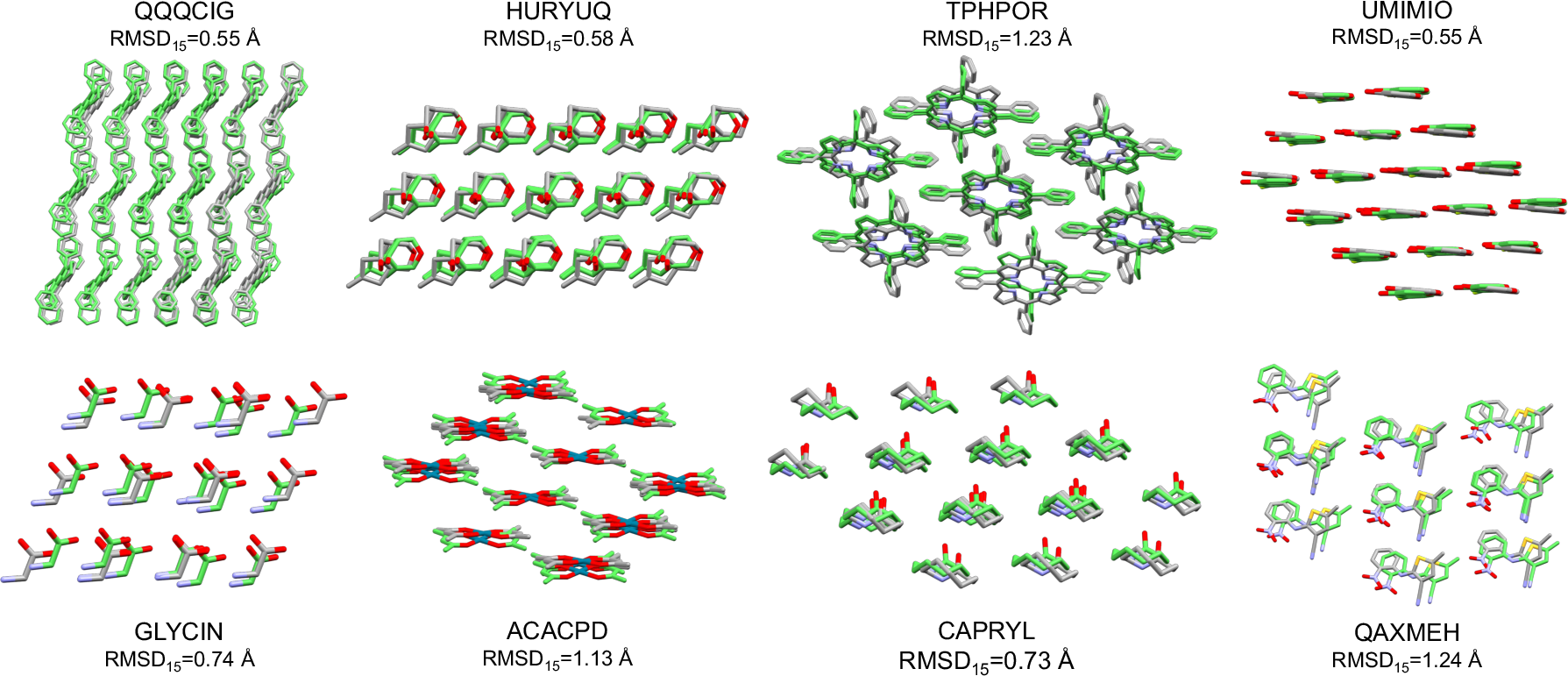
- Accuracy: OXtal often predicts molecular shapes in the crystal with very small errors (RMSD around 0.5 Å for many cases; Ångström is a tiny unit—one ten-billionth of a meter). Lower RMSD means atoms are very close to their true positions.
- Packing similarity: OXtal frequently matches how molecules pack together in real crystals, often above 80% similarity in benchmark tests.
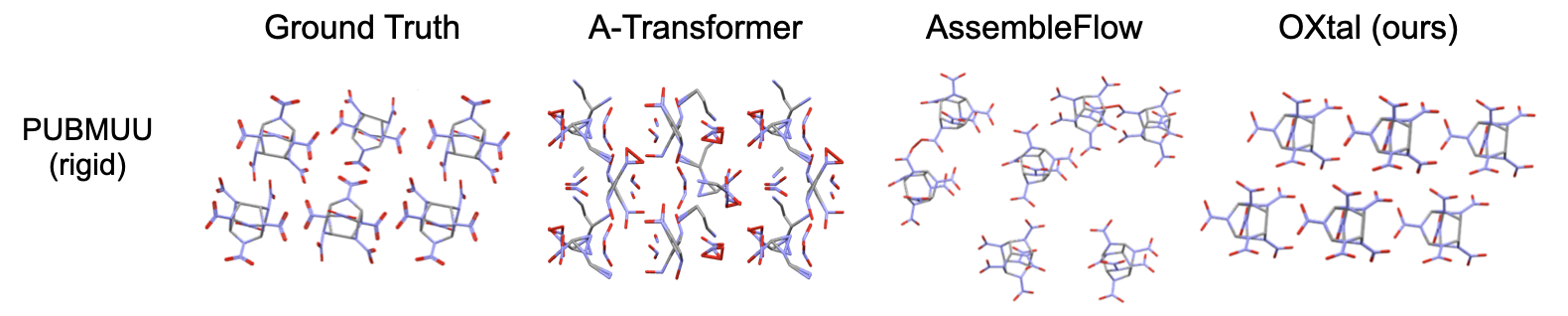
- Beats other AI baselines: Compared to other machine learning models, OXtal produces far fewer collisions (atoms overlapping unrealistically), recovers more correct molecular shapes, and matches real packing structures much more often.
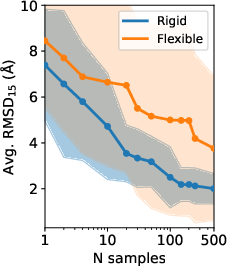
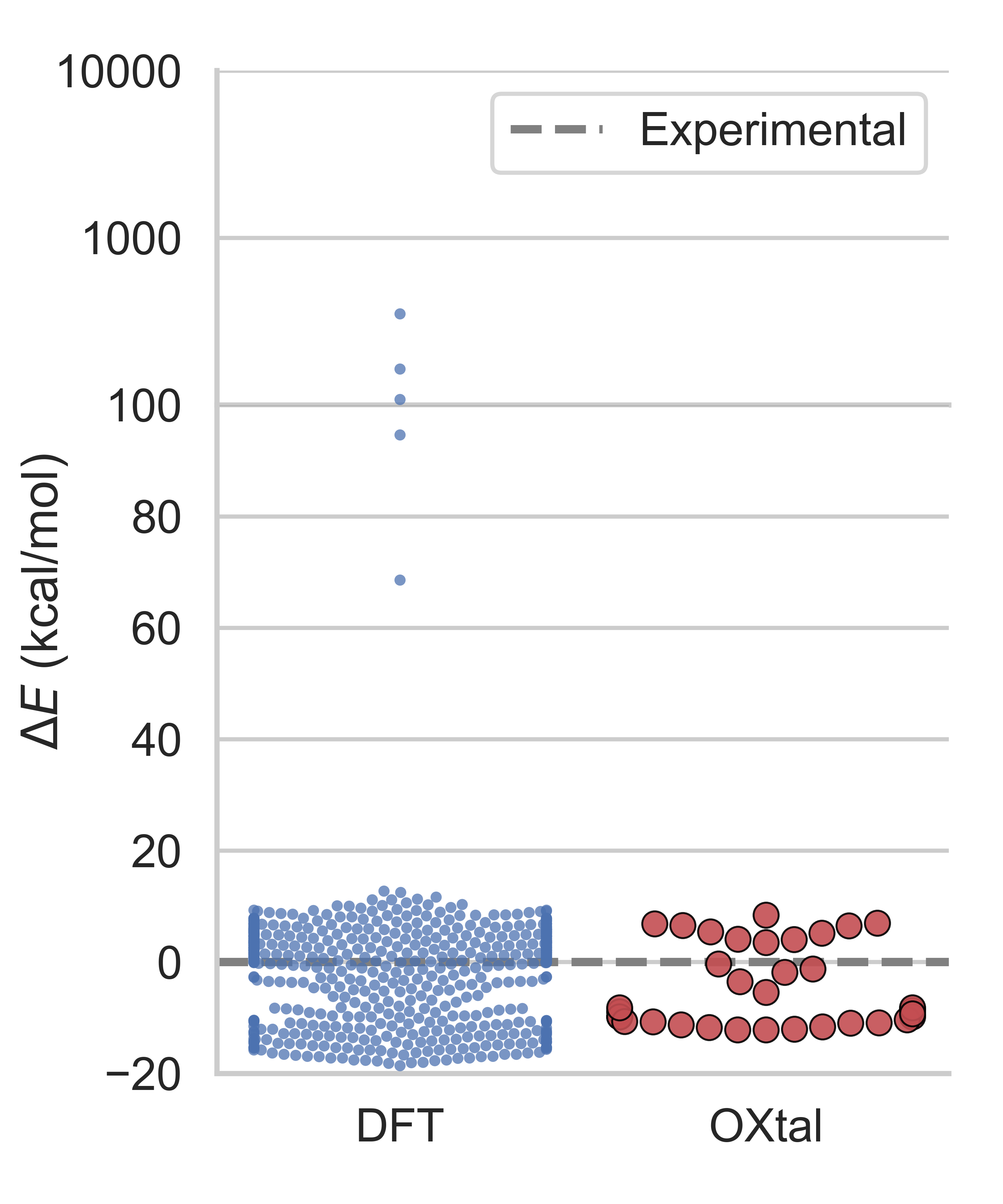
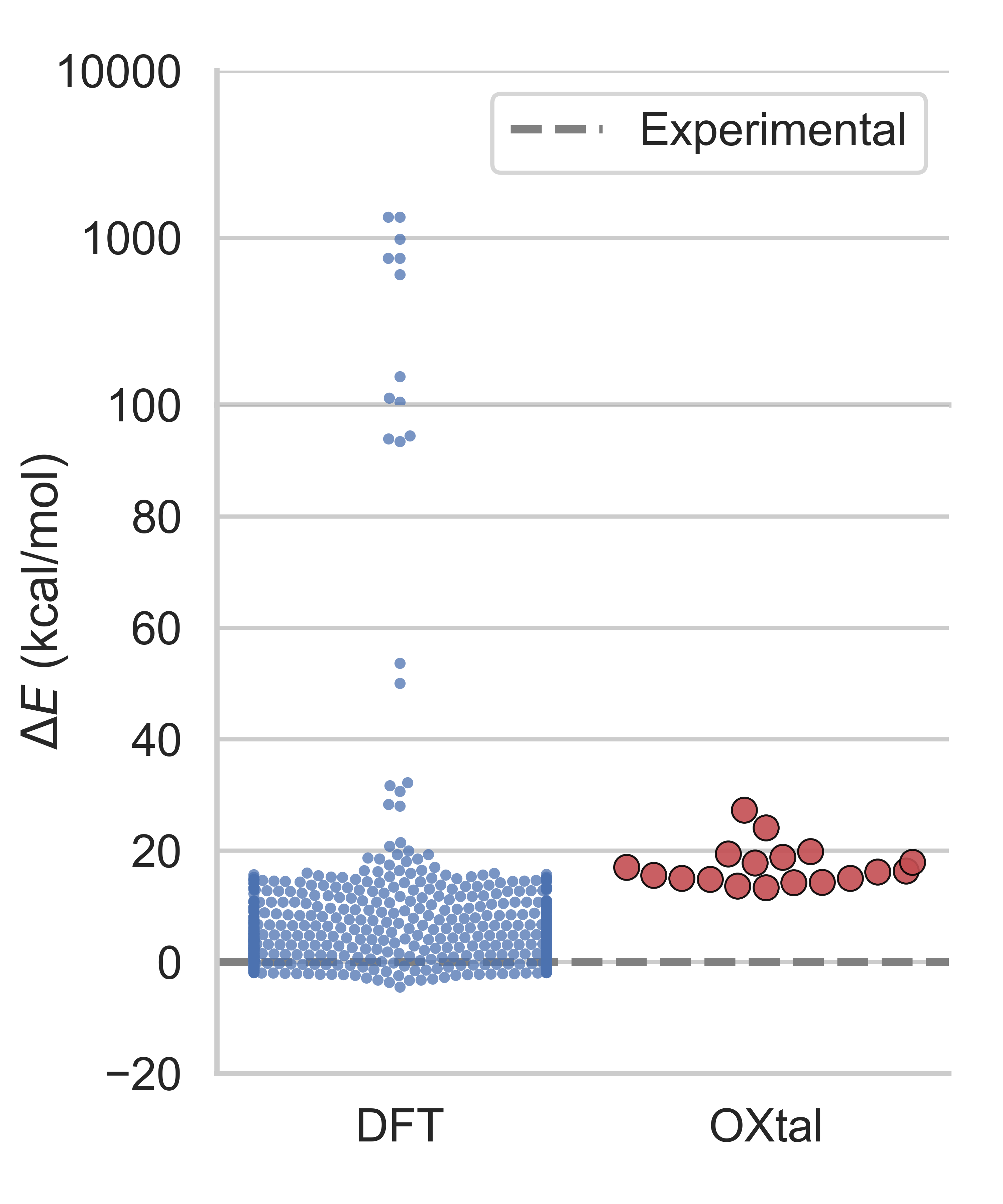
- Competitive with physics-heavy methods at a fraction of the cost: Traditional quantum chemistry simulations (like DFT) are very accurate but extremely slow and expensive, sometimes needing millions of CPU hours. OXtal reaches similar packing quality in far fewer samples and is orders of magnitude cheaper to run.
- Few-shot success: For many targets, OXtal gets close to the correct structure within just a handful of samples, making it practical for screening and design.
- Chemically sensible details: OXtal captures meaningful interactions—hydrogen bonds, halogen contacts, π–π stacking—and can even predict different known polymorphs (distinct crystal forms of the same molecule) and complex co-crystal patterns with alternating donor and acceptor molecules.
Why this matters: It suggests the model learns both the “thermodynamics” (what’s stable) and the “kinetics” (what’s likely to form under real lab conditions), not just blindly chasing energy minima.
Why does this matter?
If you can quickly predict how molecules crystallize:
- Medicines: You can foresee which crystal form will dissolve best, stay stable longer, or be more bioavailable in the body.
- Materials: You can design organic semiconductors, sensors, and batteries with better performance by choosing crystal structures that conduct charge or light more efficiently.
- Speed and cost: Instead of relying on huge numbers of expensive simulations, researchers can generate realistic candidates fast and then optionally refine with physics-based methods.
OXtal shows that large, data-driven models can learn the rules of crystal packing directly from examples. This opens the door to rapid exploration of chemical space, discovering useful materials and drug forms much faster. Future work could make it even stronger by adding smart ranking and small local relaxations, and by conditioning on lab conditions like solvent and temperature.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a single, concrete list of what remains missing, uncertain, or unexplored in the paper. Each point is phrased to enable targeted follow-up by future researchers.
- Explicit lattice prediction and evaluation: OXtal avoids unit-cell parametrization; the paper does not report accuracy for lattice parameters, fractional coordinates, space group assignment, or multiplicity Z. Define methods to recover a canonical crystallographic description from samples and evaluate against ground truth.
- Periodicity enforcement and symmetry consistency: Without an explicit lattice, it is unclear how global periodic boundary conditions and space-group symmetries are guaranteed across the infinite crystal. Develop procedures to reconstruct the full periodic structure from local samples and verify PBC consistency and symmetry operations.
- Long-range interactions under S4 cropping: The theoretical bound assumes local losses with finite interaction range, but real crystal energetics include long-range Coulomb and dispersion interactions. Quantify truncation error vs crop size for different chemotypes, and extend S4 with mechanisms (e.g., learned electrostatics, Ewald-like features, global context tokens) to capture long-range physics.
- S4 hyperparameter robustness: The impact of r_cut, token budget T_max, shell count K, and stoichiometric subsampling weights is only partially explored. Systematically ablate these across anisotropic packings, porous/low-density crystals, and highly flexible molecules to establish safe defaults and adaptive schemes.
- Kinetic modeling and conditioning: The model claims to reflect kinetic regularities but does not condition on crystallization context (solvent, temperature, supersaturation, additives). Introduce conditioning variables, train with context-annotated data, and validate by reproducing condition-dependent polymorph distributions and relative frequencies.
- Energy ranking and relaxation: OXtal does not integrate energy models or local geometry relaxation. Evaluate whether adding learned interatomic potentials or lightweight relaxations improves collision rates and solve rates; design calibrated ranking schemes tied to Gibbs free energy and kinetic accessibility.
- Protonation state, tautomers, and charge balance: Inputs assume fixed 2D graphs and charges, while many crystals involve proton transfer, salt formation, hydrates, and tautomerization. Develop joint modeling of protonation/tautomer states and counterions, or conditioning on crystallization media, and evaluate correctness of predicted charge states.
- Solvates, hydrates, and multi-component stoichiometry: Although co-crystals are demonstrated, the paper does not assess correctness of per-species stoichiometric ratios (Z′, Z) or solvent inclusion. Add metrics and generation mechanisms that predict and verify stoichiometry and solvent occupancy, including variable-component inference.
- Hydrogen placement and H-bond networks: Metrics emphasize non‑hydrogen RMSD and packing similarity. Quantitatively evaluate hydrogen positions, hydrogen-bond geometries (angles/distances), and network topology, which critically affect crystal stability.
- Space-group inference and scoring: The paper does not evaluate predicted space groups. Develop robust space-group inference from generated structures and benchmark space-group accuracy and symmetry consistency.
- Physical properties and stability checks: No results are reported for predicted densities, lattice constants, elastic/phonon stability, or PXRD agreement. Add property prediction and validation (e.g., simulated diffraction) to ensure crystallographic and thermodynamic plausibility.
- Scalability to very large unit cells: Performance on crystals with hundreds–thousands of atoms per unit cell (large peptides, host–guest frameworks, molecular cages) is not characterized. Investigate hierarchical generation or tiling strategies, memory/runtime scaling, and failure modes at extreme sizes.
- Coverage of challenging classes: There is no systematic evaluation on salts/ionic crystals, Z′>1 systems, disordered/partially occupied structures, modulated crystals, polymorph-rich APIs beyond selected examples. Create targeted benchmarks and analyze OOD generalization.
- Organometallic and coordination chemistry: The model uses standard molecular features; coordination environments in organometallics are complex. Assess whether ligand-field preferences, coordination numbers, and geometry are captured, and extend features/training if needed.
- Dataset quality and bias: CSD contains disorder, partial occupancy, and measurement artifacts. Analyze and mitigate dataset biases (space-group distribution, element coverage, stoichiometry), define cleaning protocols, and test robustness to noisy labels.
- Equivariance vs augmentation trade-off: The paper abandons explicit equivariance for SE(3) augmentation. Study whether hybrid equivariant/non‑equivariant architectures improve sample efficiency or accuracy, especially for rare symmetries or low-data regimes.
- Uncertainty quantification: There are no per-sample confidence scores or calibrated probabilities. Develop uncertainty metrics to select high-confidence predictions and quantify variability across sampler settings and seeds.
- Fairness of DFT comparisons: DFT baselines used far larger sample and compute budgets. Perform matched‑budget comparisons (including optional relaxation/ranking pipelines) and analyze complementary failure modes.
- Robustness to input conformer and feature errors: The atom encoder depends on ETKDG+xTB conformers and Mulliken charges. Quantify sensitivity to poor initial conformers/charges and explore fully graph‑based or learned charge conditioning to reduce reliance on external QC steps.
- Inference and evaluation of multiplicity Z: The method marginalizes over unknown Z but does not detail how Z is inferred at generation or evaluated. Formalize Z prediction for each species and measure accuracy relative to experimental unit cells.
- Integration with downstream refinement: Design and quantify hybrid pipelines where OXtal proposes candidates and physics-based methods refine/rank them; measure speed‑accuracy trade‑offs and recommend best practices for practical CSP workflows.
- Metrics beyond COMPACK: Packing similarity can be satisfied by partial matches. Incorporate stricter and diverse metrics (e.g., full-cell RMSD, structure factor/PXRD similarity, symmetry-consistent lattice matching) to capture crystallographic correctness.
- Reproducibility and sampling variance: Characterize convergence and variability across runs, seeds, and sampler hyperparameters; provide guidance on the number of samples needed per chemical class to reach target success rates.
- Environmental and resource footprint: Inference cost analysis is cloud‑normalized but carbon/energy usage is not quantified. Measure environmental impact, optimize for low-resource settings, and report standardized efficiency metrics.
Practical Applications
Immediate Applications
Below are applications that can be deployed now, leveraging OXtal’s demonstrated performance (RMSD₁ < 0.5 Å, >80% packing similarity within tens of samples), cost advantages over DFT, and generalization to rigid/flexible molecules, co-crystals, and solvates.
- Healthcare/Pharma — Early-stage polymorph risk mapping for APIs
- Use case: Rapidly sample and analyze plausible solid-state packings to identify potential polymorph diversity and its impact on solubility, bioavailability, and stability.
- Tools/workflows: Run OXtal (30–100 samples per molecule), align predictions using COMPACK, generate a “polymorph risk heatmap” for medicinal chemistry and CMC teams; integrate with QC/formulation planning and IP/patent strategy.
- Assumptions/dependencies: Predictions require experimental validation; OXtal is not currently conditioned on solvent/temperature; a lightweight rescoring (e.g., xTB) and ranking workflow improves reliability.
- Healthcare/Pharma — Co-crystal and salt screening for solubility/stability enhancement
- Use case: Prioritize co-formers/salts by predicted donor-acceptor interactions, hydrogen-bond networks, and packing motifs that correlate with improved solid-state properties.
- Tools/workflows: “Co-crystal recommender” feeding OXtal packings into COMPACK similarity and property heuristics (e.g., H-bond counts, packing density); shortlist candidates for lab validation.
- Assumptions/dependencies: Requires a curated co-former list; incorporate fast energy/ranking (GFN2-xTB, ML potentials) to triage; environment dependence (solvent, kinetics) still needs experimental confirmation.
- Materials/Electronics — Pre-screening organic semiconductors by packing motif
- Use case: Identify molecules likely to form target packings (e.g., π–π stacking distances, herringbone/brickwork registry) that correlate with charge transport.
- Tools/workflows: “Semiconductor packing simulator” combining OXtal predictions with charge transport estimators (e.g., Marcus rates, KMC) to rank candidates for synthesis.
- Assumptions/dependencies: Requires downstream property models and optional local relaxation; generalization strongest within small-molecule organic crystals.
- Computational Chemistry/Software — DFT warm-start and triage
- Use case: Cut the number of expensive DFT geometry optimizations by seeding with OXtal’s packing-similar structures.
- Tools/workflows: “OXtal-DFT warm-start” pipeline: OXtal sampling → collision filtering → fast xTB rescoring → select few structures for DFT refinement.
- Assumptions/dependencies: DFT still needed for final ranking; performance depends on the chemical domain and the quality of fast rescoring.
- Academia/Crystallography — Powder XRD structure solution assist
- Use case: Provide plausible starting models for Rietveld refinement and powder pattern fitting, accelerating structure solution.
- Tools/workflows: Fit experimental diffraction patterns starting from OXtal’s packing-similar candidates; use COMPACK-guided selection.
- Assumptions/dependencies: Requires alignment between predicted and experimental lattices; may need symmetry reconciliation and refinement.
- Manufacturing/Process Development — Seed selection and scale-up guidance
- Use case: Inform choice of seeding crystals and expected packing motifs to mitigate habit changes and polymorph surprises during scale-up.
- Tools/workflows: “Crystallization seed advisor” selecting seeds consistent with OXtal’s packings; integrate with process analytical technology.
- Assumptions/dependencies: OXtal does not yet condition on crystallization context; final process choices require empirical verification.
- Cheminformatics — Solid-state descriptors for property prediction
- Use case: Augment QSAR/QSPR models with OXtal-derived solid-state features (packing density, H-bond network topology, π–stack geometry).
- Tools/workflows: Feature extraction from OXtal predictions; integrate into ML property models (e.g., dissolution rate, mechanical stability).
- Assumptions/dependencies: Feature relevance depends on the endpoint; ensure model calibration against experimental data.
- Sustainability/Policy within organizations — HPC cost and energy reduction
- Use case: Replace large-scale DFT-based CSP screening with OXtal to reduce compute cost and carbon footprint while maintaining high packing similarity rates.
- Tools/workflows: Internal policy to default to OXtal for CSP triage; on-demand cloud deployment of an “OXtal-API.”
- Assumptions/dependencies: Acceptance by stakeholders; validation protocols for high-stakes decisions; ongoing monitoring for out-of-distribution chemistries.
Long-Term Applications
Below are applications that require further research, scaling, integration, or development (e.g., ranking, local relaxation, environment conditioning), but are feasible extensions of OXtal and its innovations (notably the S⁴ lattice-free training for periodic systems).
- Healthcare/Pharma/Manufacturing — Environment-conditioned crystallization planning
- Use case: Predict which polymorph forms under specific conditions (solvent, temperature, supersaturation, additives), guiding process design and scale-up.
- Tools/workflows: Extend OXtal with conditioning on crystallization context; integrate with self-driving labs for closed-loop optimization.
- Assumptions/dependencies: Requires curated, condition-labeled training data; robust ranking/relaxation; real-time lab instrumentation.
- Autonomous Labs/Robotics — Closed-loop polymorph control via active learning
- Use case: Actively explore and fix polymorph outcomes using OXtal suggestions, rapid experiments, and updated models.
- Tools/workflows: On-line Bayesian optimization over crystallization parameters; OXtal-generated candidates; automated analytics.
- Assumptions/dependencies: Reliable feedback from high-throughput crystallization; standardized data formats and lab automation.
- Regulatory/Policy — Standardized computational polymorph risk assessment
- Use case: Formalize OXtal-based workflows (with physics-based verification) as part of ICH Q6A and FDA/EMA guidance for polymorphism.
- Tools/workflows: Validation studies across therapeutic classes; shared benchmarks and acceptance criteria.
- Assumptions/dependencies: Broad community acceptance; reproducibility and auditability; documented uncertainty quantification.
- Materials/Energy/Electronics — Property-driven inverse design of molecular crystals
- Use case: Jointly optimize molecules and packings for target properties (charge mobility, porosity, optical response, mechanical robustness).
- Tools/workflows: OXtal + property predictors + generative molecular design; multi-objective search; physics-informed filters.
- Assumptions/dependencies: Differentiable or surrogate property models; scalable search and ranking; domain-appropriate datasets.
- Expanded domains — Polymer crystals, biomolecular crystals, MOFs/COFs-like organic frameworks
- Use case: Adapt S⁴ and non-equivariant large transformers to other periodic and mixed systems with larger unit cells or complex symmetries.
- Tools/workflows: Domain-specific data curation; hybrid representations (molecular + framework topology); symmetry reconciliation tools.
- Assumptions/dependencies: Adequate labeled datasets; architectural and training modifications; evaluation metrics aligned to each domain.
- CSP-as-a-Service — Industrial-scale cloud platforms for CSP and crystal engineering
- Use case: Centralized, secure platforms offering OXtal-based CSP, ranking, and process design modules to pharma and materials companies.
- Tools/workflows: APIs, compliance and audit trails, integration with ELNs and LIMS; cost-aware sampling and scheduling.
- Assumptions/dependencies: Productization, security and IP considerations; SLAs and validation frameworks.
- Near-DFT single-shot prediction — Integrated ranking and local relaxation
- Use case: Achieve DFT-level accuracy in a few shots by coupling OXtal with learned energy models and rapid local relaxations.
- Tools/workflows: ML interatomic potentials, physics-informed refiners; uncertainty-aware ranking.
- Assumptions/dependencies: High-quality energy models across diverse chemistries; calibrated uncertainty estimates.
- Sustainability/Policy at scale — HPC energy reduction and benchmarking standards
- Use case: Sector-wide replacement of brute-force DFT CSP campaigns with OXtal-hybrid workflows to cut energy use and cost.
- Tools/workflows: Shared benchmarks and reporting (compute hours, carbon metrics); procurement guidelines favoring efficient methods.
- Assumptions/dependencies: Stakeholder buy-in; transparent audits; continuous performance tracking.
- Legal/IP Analytics — Predictive polymorph landscape for patent strategy
- Use case: Inform patent filings and freedom-to-operate analyses by mapping plausible crystal forms computationally.
- Tools/workflows: OXtal-generated polymorph ensembles + COMPACK similarity + prior art databases; risk scoring.
- Assumptions/dependencies: Judicial/regulatory acceptance; clear documentation of methods and limitations.
- Safety/Defense — Energetic materials risk screening
- Use case: Evaluate packing-induced sensitivity risks prior to synthesis, guiding safer materials development.
- Tools/workflows: Structure-based risk models fed by OXtal packings; prioritize safer candidates for testing.
- Assumptions/dependencies: Validated structure–risk relationships; careful domain adaptation and expert oversight.
Glossary
- ab initio: From first principles without empirical parameters, typically referring to physics-based calculations. "ab initio molecular crystal structure prediction (CSP) seeks to estimate the distribution of experimentally realizable crystal packings in an accurate and scalable manner."
- asymmetric unit: The minimal subset of atoms that, under the crystal’s symmetry operations, generates the full unit cell. "A periodic crystal admits an asymmetric unit , the minimal subset that recovers the entire unit cell by applying symmetry transformations of the crystal's space group."
- Bregman divergence: A family of distance-like measures derived from a convex function, used in optimization and learning objectives. "any Bregman divergence with convex "
- Cambridge Structural Database (CSD): A large curated repository of experimentally determined crystal structures. "We next curate a training dataset from the Cambridge Structural Database (CSD) that contains k crystals."
- Cartesian coordinates: Atom positions expressed in standard Euclidean space (x, y, z), as opposed to fractional coordinates. "or equivalently, its Cartesian coordinates ."
- co-crystal: A crystalline solid composed of two or more different molecular species in a defined stoichiometric ratio. "including rigid and flexible molecules, co-crystals, and solvates"
- conformer: A specific 3D arrangement of a molecule due to rotation around single bonds. "conformer Å"
- COMPACK: A CSD tool for comparing crystal packings by aligning molecular clusters. "Using CSD COMPACK, a sample's packing is partially similar if at least 8 of 15 molecules could be aligned to an experimental cluster"
- density functional theory (DFT): A quantum-chemical method that computes electronic structure based on electron density. "force fields or quantum-chemical density functional theory (DFT)"
- diffusion transformer: A transformer-based neural architecture used within diffusion models to predict denoised outputs. "a large 70M parameter diffusion transformer"
- distogram: A binned representation of pairwise distances, often used as a model output or loss target. "we also include a distogram loss on a separate head branching from the trunk"
- Evoformer: An equivariant neural network block from AlphaFold2 that processes sequence and pairwise features for structure prediction. "Unlike AlphaFold2, which relied on the equivariant Evoformer \citep{af2} architecture"
- fractional coordinates: Atom positions expressed relative to the lattice vectors within the unit cell. "its fractional coordinates relative to the lattice vectors"
- GFN2-xTB: A semi-empirical quantum chemical method used for geometry relaxation and energy evaluation. "relaxation by the semi-empirical quantum chemical method GFN2-xTB"
- Gibbs free energy: A thermodynamic potential determining stability under constant temperature and pressure. " is the Gibbs free energy (thermodynamics)"
- Itô stochastic differential equation (SDE): A formulation of SDEs under Itô calculus, describing stochastic dynamics. "a (It^o) stochastic differential equation (SDE)"
- lattice vectors: The three vectors defining the periodic translation basis of a crystal’s unit cell. "defines the lattice vectors forming a parallelepiped known as the unit cell."
- minimum-image intermolecular distance: The shortest distance between molecules accounting for periodic boundary conditions. "We next define the minimum-image intermolecular distance between two molecules, ."
- Mulliken partial charges: Atomic charges derived from Mulliken population analysis of electronic structure. "Mulliken partial charges"
- nucleation: The initial formation of an ordered small cluster that seeds crystal growth. "nucleation and growth pathways"
- Pairformer: A triangular attention-based module (from AlphaFold3) updating atom-level single and pair representations. "We then apply the Pairformer stack from AlphaFold3"
- polymorph: A different crystal packing arrangement of the same chemical substance. "including crystal polymorphs, and generalize to complex co-crystal and biomolecular interactions"
- RDKit ETKDG: An algorithm combining experimental torsion knowledge with distance geometry to generate 3D conformers. "we generate a conformer with RDKit ETKDG"
- RMSD1: Root-mean-square deviation computed on one molecule’s non-hydrogen atoms; used to assess conformer recovery. "RMSD\textsubscript{1}\,\AA{}"
- RMSD15: Root-mean-square deviation computed over a 15-molecule cluster to assess packing accuracy. "RMSD\textsubscript{15}\,\AA{} on a 15-molecule cluster."
- SE(3): The group of 3D rigid-body transformations (rotations and translations), encoding geometric symmetries. "employing data augmentation."
- sLDDT: Smooth local distance difference test; a differentiable metric assessing local structural accuracy. "a smooth local difference distance test $\mathcal{L}_{\text{sLDDT}$ as defined in \citet{af3}"
- space group: The set of symmetry operations (translations, rotations, reflections, glide planes, etc.) that define a crystal’s symmetry. "symmetry transformations of the crystal's space group."
- Stein score: The gradient of the log-density, used to construct the reverse-time dynamics in diffusion models. "linked via the Stein score "
- stoichiometric ratio: The relative counts of different components in a multi-component crystal. "co-crystal polymorphs with 1:1 and 2:1 stoichiometric ratio."
- Stoichiometric Stochastic Shell Sampling (S4): A lattice-free training scheme that samples concentric shells while preserving component ratios. "Stoichiometric Stochastic Shell Sampling (), a novel lattice-free training scheme"
- supercell: An enlarged cell formed by integer combinations of lattice vectors that tiles the same infinite crystal. "A supercell can be obtained by an integer matrix "
- unit cell: The smallest repeating parallelepiped that defines the periodic structure of a crystal. "forming a parallelepiped known as the unit cell."
- van der Waals radii: Empirical radii representing weak non-bonded contact distances between atoms. "where is the sum of atomic van der Waals radii"
- Wiener process: Standard Brownian motion used as the stochastic term in SDEs. "the diffusion coefficient for the Wiener process $#1{W}_t$."
Collections
Sign up for free to add this paper to one or more collections.