- The paper demonstrates that pretraining data diversity drives superior viral classification and generative performance over mere parameter scaling.
- The paper reveals significant performance degradation in NFMs under genus-disjoint and temporal splits, highlighting challenges in evolutionary generalization.
- The paper shows that traditional likelihood metrics fail to capture biological sequence fidelity, urging the adoption of more biologically grounded evaluation methods.
ViroBench: A Comprehensive Benchmark for Nucleotide Foundation Models in Viral Genomics
ViroBench presents a systematic framework for evaluating nucleotide foundation models (NFMs) on viral genomics applications. It addresses critical needs in bioinformatics by unifying assessment of both discriminative biological understanding and generative, biosecurity-relevant behavior of NFMs across diverse virus lineages, tasks, and evolutionary regimes. This essay provides an in-depth, technical discussion of the design, analysis, and major findings from ViroBench (2605.25388), emphasizing implications for foundation model development in biological sequence modeling and biosecurity risk assessment.
Benchmark Construction and Experimental Design
ViroBench is architected as a diagnostic, multidimensional benchmark for NFMs in virology, encompassing 58,314 high-quality viral sequences, comprehensive taxonomy and host annotation, and controlled evaluation regimes that reflect realistic phylogenetic and temporal shifts (Figure 1).
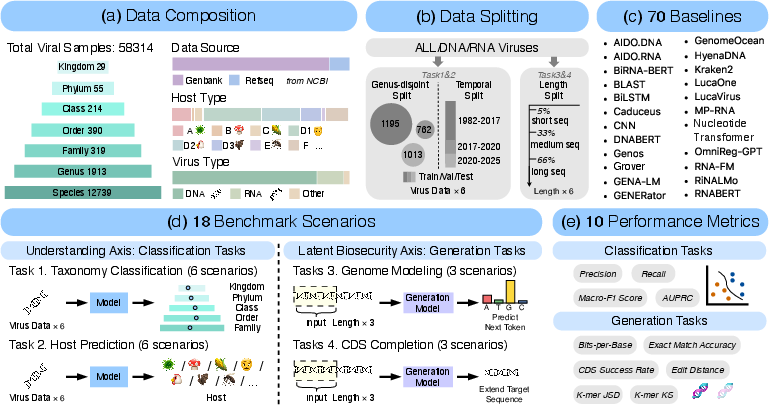
Figure 1: ViroBench design provides diversity in data composition, splitting protocols, baseline model coverage, scenario types, and evaluation metrics.
Data curation starts from NCBI viral TaxIDs and integrates hierarchical taxonomy, standardized host types, and temporal metadata for rigorous biological grounding. Host labels are resolved using LLM-powered normalization, yielding robust, coarse-grained groupings critical for downstream classification and risk analysis. Filtering steps enforce both sequence quality and metadata completeness.
ViroBench instantiates two principal diagnostic axes:
- Biological Understanding: 12 classification scenarios, including taxonomy and host prediction, are designed to probe the depth of viral rule acquisition by NFMs. Two rigorous splits are enforced:
- Genus-disjoint split: Ensures strict phylogenetic generalization and guards against shallow sequence memorization.
- Temporal split: Simulates mutational drift, testing extrapolation to contemporaneous and future viral variants.
- Latent Biosecurity Risk: Six generative scenarios systematically examine sequence likelihood modeling and functional coding sequence (CDS) generation. Evaluation is stratified by sequence length, probing the stability of models over longer generative trajectories relevant for synthetic biology risk control.
The full suite includes 66 NFMs, spanning diverse architectural paradigms (BERT, Transformers, SSMs, Hyena variants) and pre-training regimes (general, phage-specific, RNA-specific, and non-viral coverage), supplemented by four conventional baselines.
Evaluating Biological Understanding
Classification Performance Drivers
The benchmark reveals that pretraining data coverage and taxonomic diversity, rather than parameter scaling alone, are the dominant factors for robust viral understanding. Models such as LucaVirus and Evo2-40B achieve high classification Macro-F1 by leveraging diverse viral exposure and, in some cases, extreme scale. Interestingly, parameter scaling allows non-viral models (e.g., AIDO.DNA-7B) to extract latent viral patterns from metagenomic fragments despite an absence of viral sequences in training, indicating information transfer via endogenous viral elements in eukaryotic hosts.
Vulnerabilities in Extrapolation
NFMs display marked performance degradation under both genus-disjoint and temporal splits, with Genos-10B and others exhibiting catastrophic collapse in host prediction under even modest temporal drift. This underscores the current models' fragility for true real-world generalization and their sensitivity to rapid viral evolution—a central challenge for any model intended for deployment in genomic surveillance.
Phylogenetic Resolution Limitations
Model analysis via confusion matrices mapped to phylogenetic trees demonstrates that misclassifications are not randomly distributed, but are concentrated at "gray zones"—proximal clades or families in the evolutionary landscape. This pattern evidences the models' acquisition of only coarse-grained evolutionary structure, lacking sufficient resolution for fine-grained pathogen delineation (Figure below).
(Figure 2)
Figure 2: Family-level confusion matrix projected onto a phylogenetic tree; misclassifications are clustered in adjacent lineages, revealing limited resolution across deep evolutionary splits.
Decoupling of Statistical Fit and Biological Validity
The generative benchmark surfaces a critical risk: There is a substantial decoupling between per-base statistical likelihood (BPB) and biological functional validity (CDS/coding integrity and k-mer spectrum match). Some models achieve strong BPB, indicating high next-token accuracy, yet fail to maintain k-mer distributions or coding validity, thereby generating biologically implausible, potentially biased sequences. This highlights that perplexity-centric evaluation is an inadequate proxy for functional sequence fidelity in biosecurity contexts.
Length-Dependent and Host-Dependent Risk Profiles
Via explicit length bucket stratification, the benchmark shows that local compositional constraints (e.g., codon usage, motif structure) are increasingly difficult to enforce as sequence lengthens. Host-stratified evaluations using metrics such as k-mer JSD reveal differential risk windows across ecological categories, suggesting certain host groups (particularly phage- and plant-associated viruses) may represent higher or lower generative stability under current architectures.
Structural Evaluation with AlphaFold3
Testing generated CDS continuations with AlphaFold3 exposes the rarity of structural congruence between generated and reference proteins. High TM-scores (≥0.5) are observed only for a very limited fraction of targets (mainly short, phage-associated proteins), emphasizing the gap between sequence generation and fold-level biological viability.
(Figure 3)
Figure 3: Overlaid structures predicted by AlphaFold3 reveal that only a minority of generated continuations maintain fold similarity to ground-truth viral proteins.
Ablation Studies: Data Diversity vs. Model Scale
Controlled ablation experiments show taxonomic diversity in pretraining outweighs increases in parameter count for downstream viral classification and generative tasks. A lightweight Hyena variant trained on a virus-enriched, taxonomically diverse in-domain corpus (ViroBland) outperforms the original model by 67.5% Macro-F1. This effect is confirmed to be architecture-agnostic through additional re-pretraining of DNABERT2 and Caduceus models.
Practical and Theoretical Implications
The diagnostic framework and results from ViroBench suggest several major implications:
- Design of Pretraining Data: For high-fidelity viral genomics, diverse and balanced taxonomic composition is essential; naive parameter scaling is secondary for performance ceiling.
- Benchmarking Protocols: Realistic evolutionary and phylogenetic splits are necessary to uncover generalization vulnerabilities that would otherwise be hidden under random splits. Future virus-aware foundation models should prefer benchmarks like ViroBench to avoid overfitting to non-diagnostic tasks or datasets.
- Metrics for Biosecurity: Likelihood metrics (BPB/perplexity) are not reliable proxies for biosafety; models should be evaluated against biologically grounded criteria such as k-mer distributional fit, CDS functional success, and protein structure preservation.
- Limitations of Current Models: Despite advances, present NFMs still fail to guarantee sample-wise biological fidelity in both discriminative and generative tasks, highlighting significant open challenges for trustworthy application in sensitive domains such as synthetic virology and pathogen surveillance.
Conclusion
ViroBench establishes the first rigorous standardized platform for quantitative, interpretable, and reproducible benchmarking of nucleotide foundation models in viral genomics. Its protocol surfaces previously masked vulnerabilities of both classification and generative architectures under realistic evolutionary shifts. By demonstrating the outsized benefit of taxonomic data diversity over model scaling and highlighting key disconnects between likelihood fit and biological validity, ViroBench provides an essential tool for the community. Future direction includes extending to multi-label and recombination-aware settings, integrating molecular-clock dating, and incorporating structure-based objectives into training. The framework and results will accelerate model design for resilient, safe, and interpretable viral sequence modeling.