- The paper introduces REQWIEM, an oracle-free quantum algorithm that leverages parallel Trotterization to simulate nonadiabatic quantum molecular dynamics without iterative oracles.
- The methodology employs grid-based split-operator propagation with rigorous error analysis to handle chaotic dynamics and strong electronic couplings.
- Empirical validations on pyrazine and Landau-Zener models confirm REQWIEM’s efficiency and scalability, paving the way for advanced quantum simulations.
Essay: An Oracle-Free Quantum Algorithm for Nonadiabatic Quantum Molecular Dynamics
Introduction and Motivation
Simulating nonadiabatic quantum molecular dynamics (NA-QMD)—where nuclear motion and multiple electronic states are entangled—remains a classically intractable problem due to an exponential scaling that arises from chaotic Hamiltonians and strong coupling, especially in the vicinity of conical intersections. Classical methodologies (e.g., ML-MCTDH, TD-DMRG, TD-CC, AIMS) offer optimized solutions for special molecular regimes or small system sizes, but are fundamentally limited by the curse of dimensionality when confronted with strongly correlated, high-dimensional, or low-symmetry molecules.
Quantum algorithms theoretically promise polynomial scaling for problems in the BQP class, with NA-QMD included. However, most existing quantum approaches—particularly those based on quantum signal processing (QSP) and block encoding—are based on oracle models that fail to scale efficiently for dense, information-rich, and unstructured Hamiltonians that are characteristic of NA-QMD, especially within the diabatic representation. These limitations are exacerbated by chaotic system dynamics, which impose severe lower bounds on the achievable circuit depth and resource requirements, eliminating the possibility of algorithmic fast-forwarding.
The work introduces and develops the REQWIEM (Real-time Evolution of Quantum Wavepackets In Explicit Modularity) algorithm, a fully explicit, oracle-free, product-formula-based quantum simulation architecture. REQWIEM leverages first-quantized, grid-based split-operator propagation, avoids expensive serial oracles in favor of parallelism and structural decomposition, and is validated with empirical observables such as recurrence spectra and population dynamics for strongly nonadiabatic systems.
NA-QMD sits within bounded-error quantum polynomial (BQP) complexity. However, the practical quantum simulation of NA-QMD is fundamentally constrained by several rigorous, entangled factors:
- Kinetic Entropy vs. Circuit/Positional Entropy: For unstructured (non-sparse) Hamiltonians in the diabatic basis, the number of independent Hamiltonian coefficients is exponential in the number of modes, demanding O(M2Nd) resources for M electronic states and d modes with N grid points. Any asymptotically optimal simulation cannot compress this "kinetic" entropy; efficient simulation must distribute it, not serialize it.
- Chaos and Non-Fast-Forwardability: Generic NA-QMD Hamiltonians, particularly near conical intersections, generate chaotic trajectories in the unitary manifold, yielding negative geodesic curvature and Lyapunov exponents that scale with system complexity. The Atia-Aharonov theorem formalizes that quantum circuits simulating such systems cannot be fast-forwarded beyond Ω(t) circuit depth. Scrambling phenomena impose an unavoidable resource overhead linear in simulation time.
- Trotterization Optimality: For noncommuting, dense Hamiltonians, product formulas (Trotterization) are proven to be as efficient, asymptotically, as polynomial oracle/query-based methods. However, the multiplication of serial oracle depths in QSP-based schemes renders them intractable for large, information-rich NA-QMD simulations.
This motivates the REQWIEM design: direct, parallelized application of Hamiltonian terms, with error bounds governed by Trotter decomposition, and resource costs that, while still exponential in modes for general systems, are amortized by circuit parallelism rather than serialized data structures.
Circuit Construction and Algorithmic Architecture
Wavefunction Preparation
The initial molecular wavefunctions—often Gaussian or other physically relevant packets—are prepared using a classically optimized, gate-based, factorized unitary coupled-cluster (UCC) ansatz, supporting up to n2 variational parameters for n-qubit registers. Classical optimization (combining Adam, warm-restart basin hopping, L-BFGS, and analytical gradients via parameter-shift) reliably achieves ∼10−11 infidelity for n≤12.
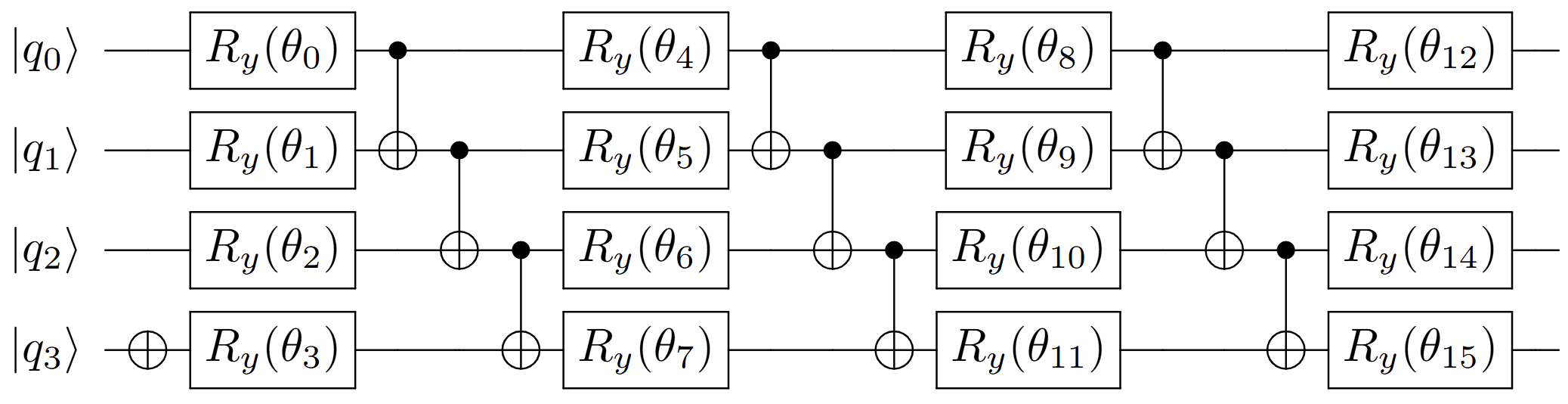
Figure 1: Factorized unitary coupled-cluster ansatz circuit for n=4 qubits. Classical emulation can optimize on the ground state of a harmonic oscillator, or maximize state fidelity with a classically-initialized function.
Grid Discretization and Error Scaling
Wavefunction propagation uses binary encoding for each degree of freedom. Rigorous error analysis, connecting grid size M0, physical energy cutoffs and Trotterization error, establishes constraints for M1 to achieve chemical accuracy and avoid aliasing. Temporal error accumulation and spectral norm scaling (Nyquist limits) are explicitly incorporated for robust error budgeting.
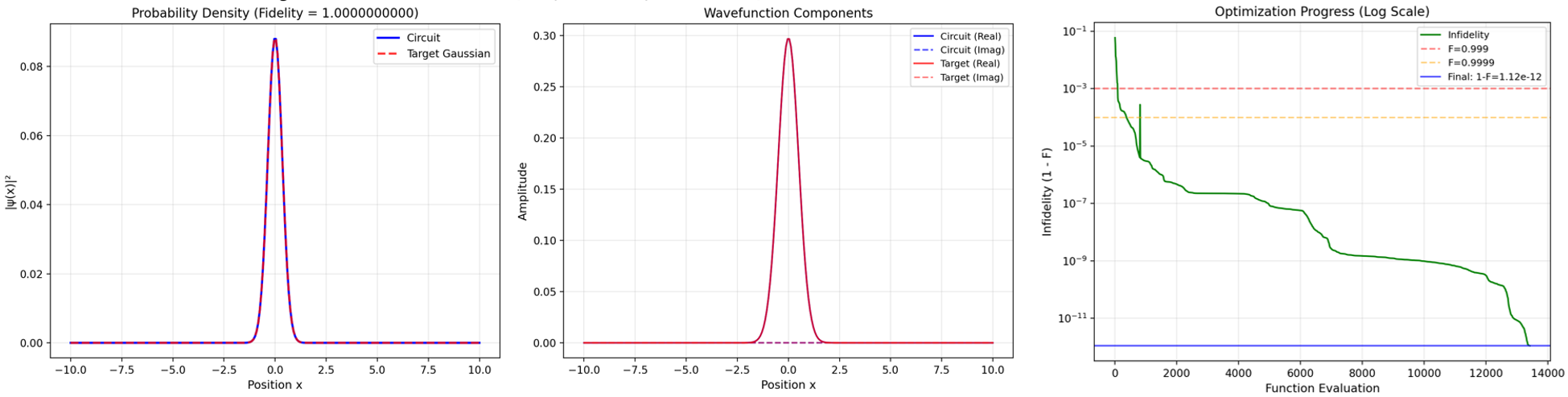
Figure 2: Gaussian wavepacket optimization on M2 qubits, showing rapid convergence of the classical optimizer and fidelity scaling for required grid sizes and error thresholds.
Simulation of NA-QMD is performed via symmetric (second-order) Trotterization: M3
with M4. Trotter error analysis is performed by explicit commutator expansion, with grid-dependent constants for both diagonal and bilinear terms, and resource-optimal parameterization for high-fidelity evolution.
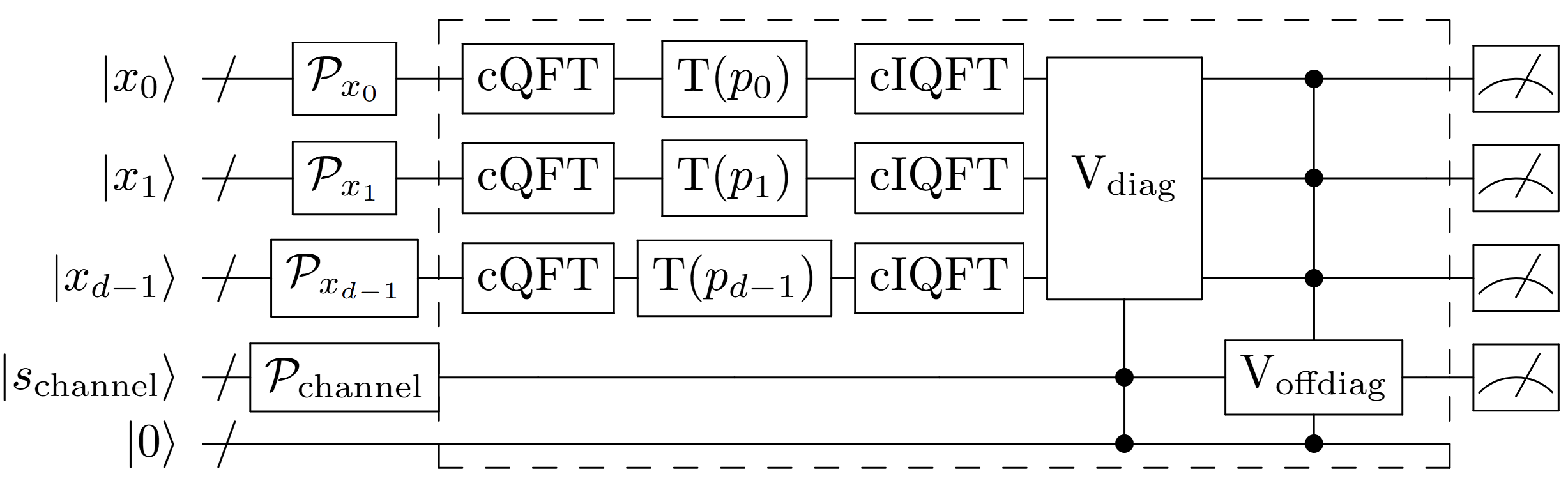
Figure 3: Schematic quantum circuit for first-order Trotterization. In this schematic, control operations depend on potential terms and do not require controls on all qubits. Registers M5 represent coordinates, each with M6 qubits. Registers M7 index electronic channels, with a work ancilla initialized to M8. Operations inside the dashed box are iterated for M9 Trotter steps.
Gate-Efficient Hamiltonian Implementation
- Kinetic Operator: Applied between QFTs, using binary-weighted d0 and d1 gates, minimizing commutator structures via tensor product commutativity.
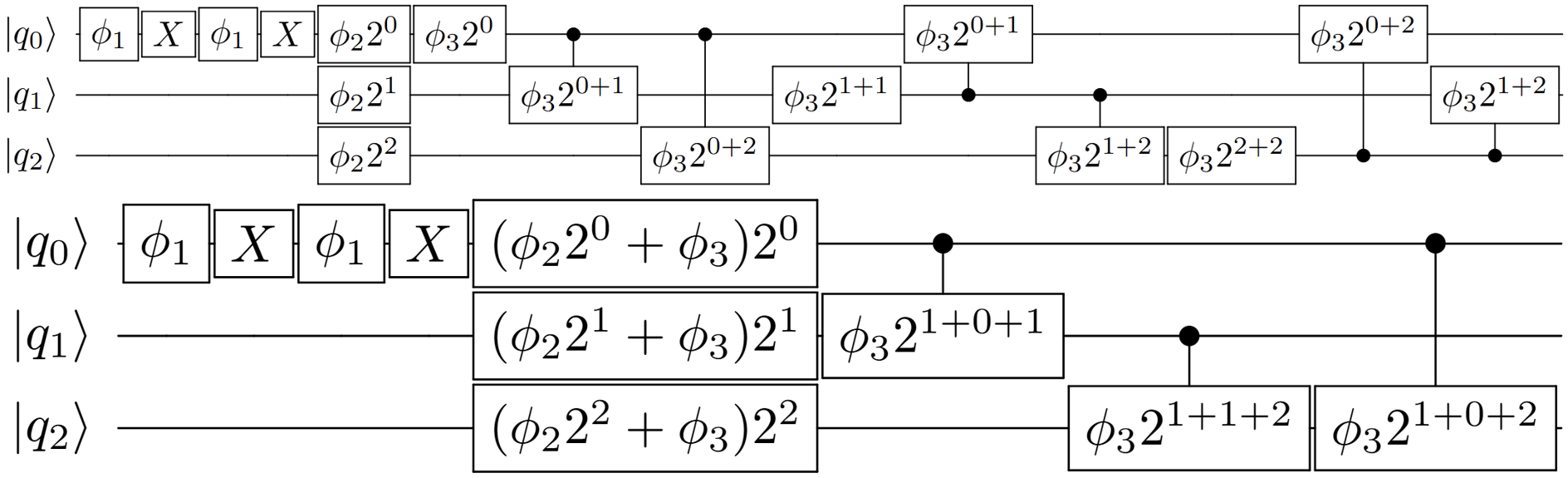
Figure 4: Explicit gate decomposition of quadratic kinetic function. Gates are grouped as global phase, first-order, and quadratic terms, weighted by the binary encoding of each qubit.
- Diagonal (On-Diagonal) Potential Terms: Multi-mode, multi-channel terms are implemented via quantum multiplexing and Gray-code uniformly controlled rotations (UCRs), leveraging molecular symmetry for gate count reduction and parallelization over modes.
- Off-Diagonal Couplings: Hamiltonian is partitioned into XOR-classes, and fragmented so each is block-diagonalized by a lightweight Clifford circuit. This supports parallel, channel-selective couplings for arbitrary d2 electronic states, leveraging fanned-out channel ancilla to enable per-mode parallelization, with bilinear terms optimized by chromatic index scheduling.
Circuit Parallelization
REQWIEM's explicit fan-out architecture supports depth scaling d3 where d4 is the chromatic index of the conflict graph of allowed bilinear couplings. This enables up to an d5-fold reduction in circuit depth for d6 modes, relative to accumulator-based (serial) architectures.
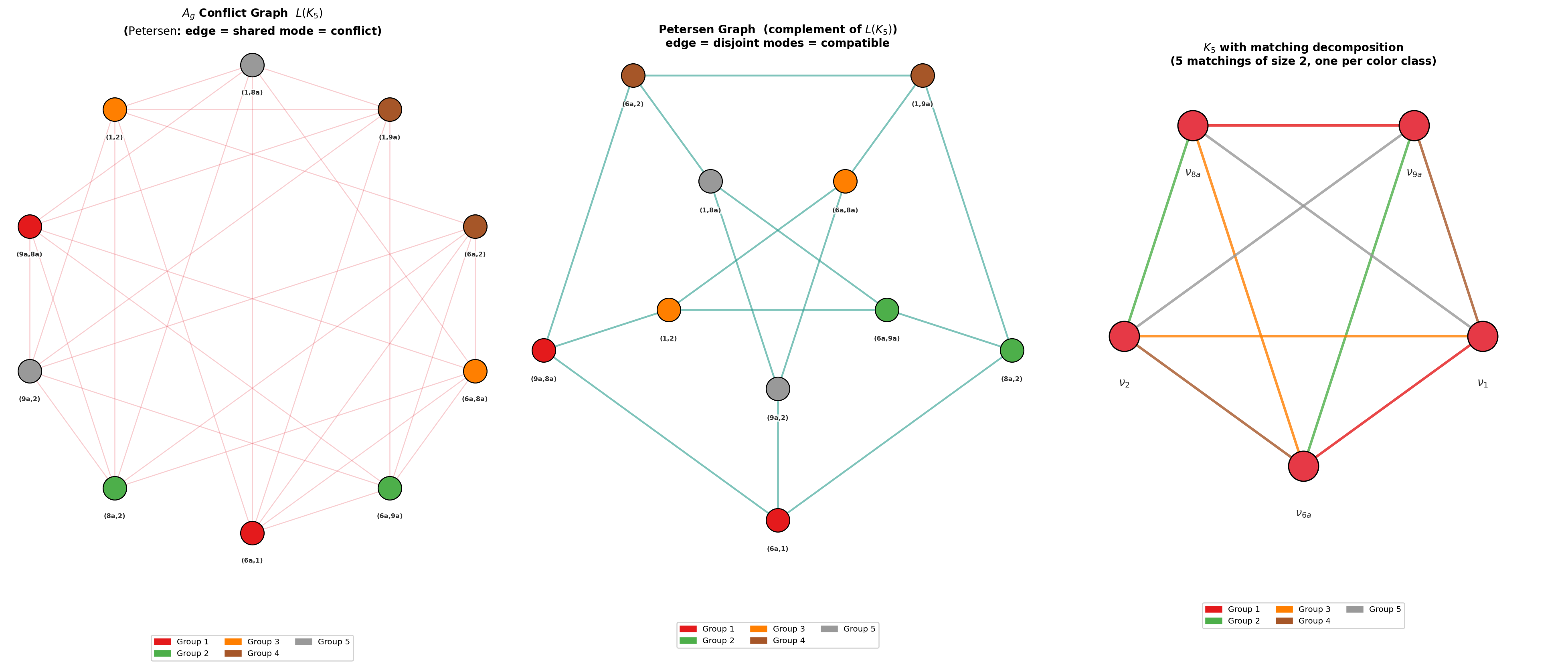
Figure 5: Conflict graph for symmetry-allowed (e.g., d7) bilinear mode pairs, illustrating how chromatic coloring enables maximal circuit parallelism for multi-dimensional simulation.
Numerical Validation and Quantum Emulation
REQWIEM is demonstrated and numerically validated via statevector emulation for prototypical NA-QMD systems:
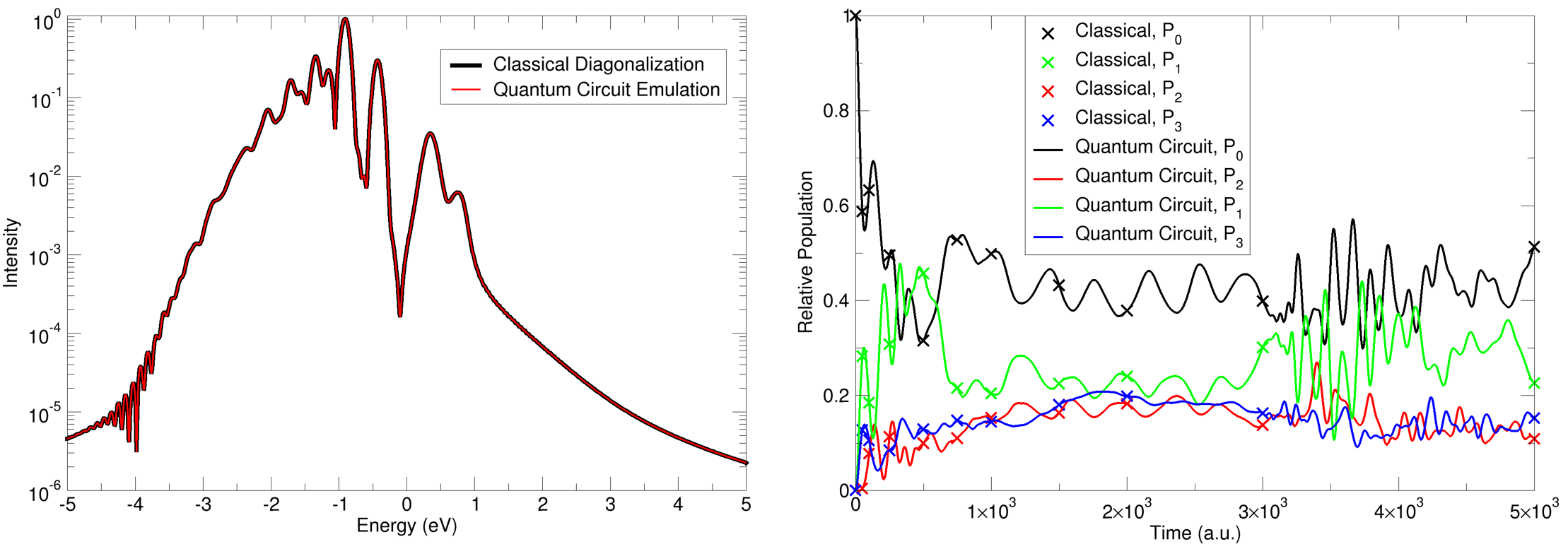
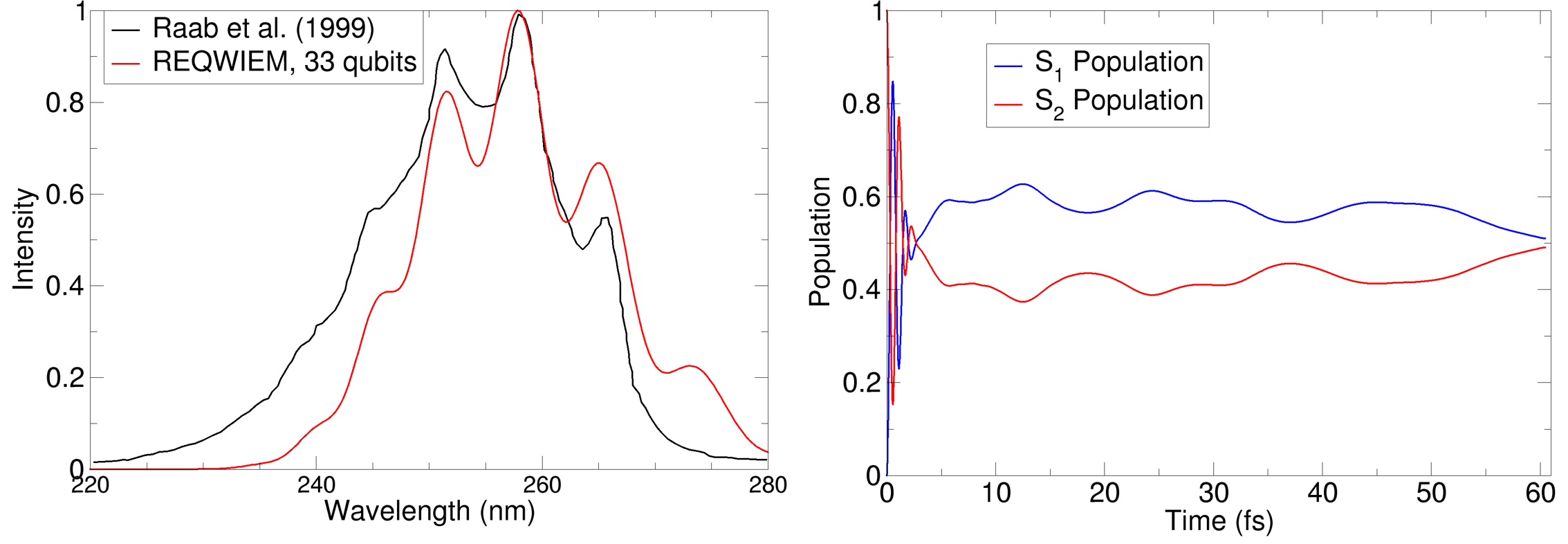
- Two- and Four-Mode Pyrazine: Simulation of ultrafast population transfer and absorption spectra in pyrazine reproduces key features of quantum recurrence, Berry phase buildup, and population relaxation, matching results from converged MCTDH calculations. Deployment on GPU clusters (multi-GPU cuQuantum) demonstrates practical scalability and resource requirements for systems up to 33 qubits.
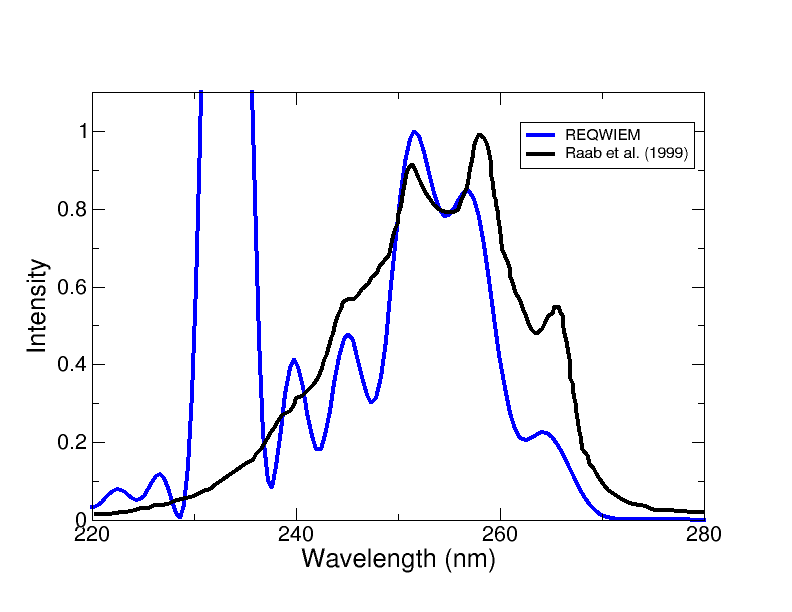
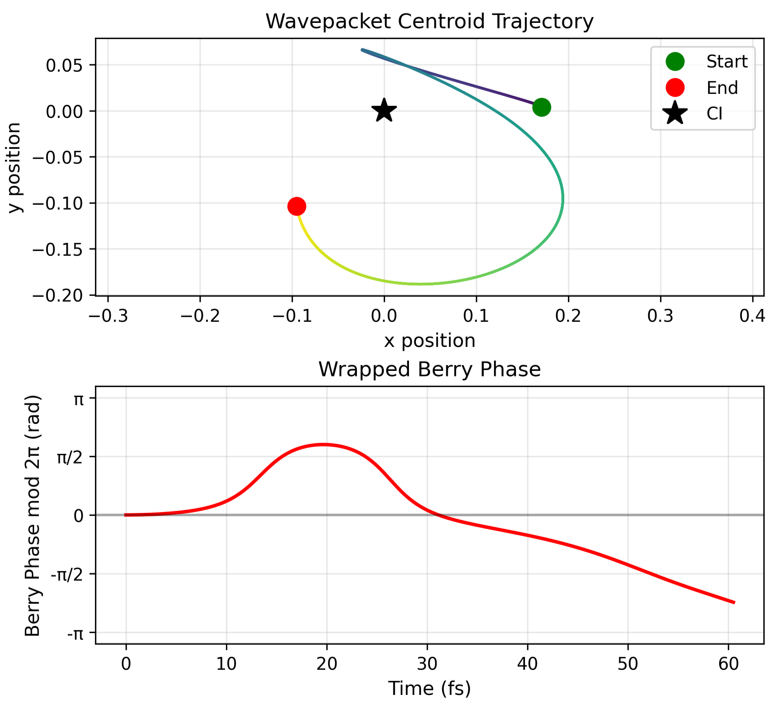
Figure 6: Spectra comparison for two-mode pyrazine simulation using REQWIEM (blue) against MCTDH calculations (black), confirming quantitative agreement in the main absorption features.
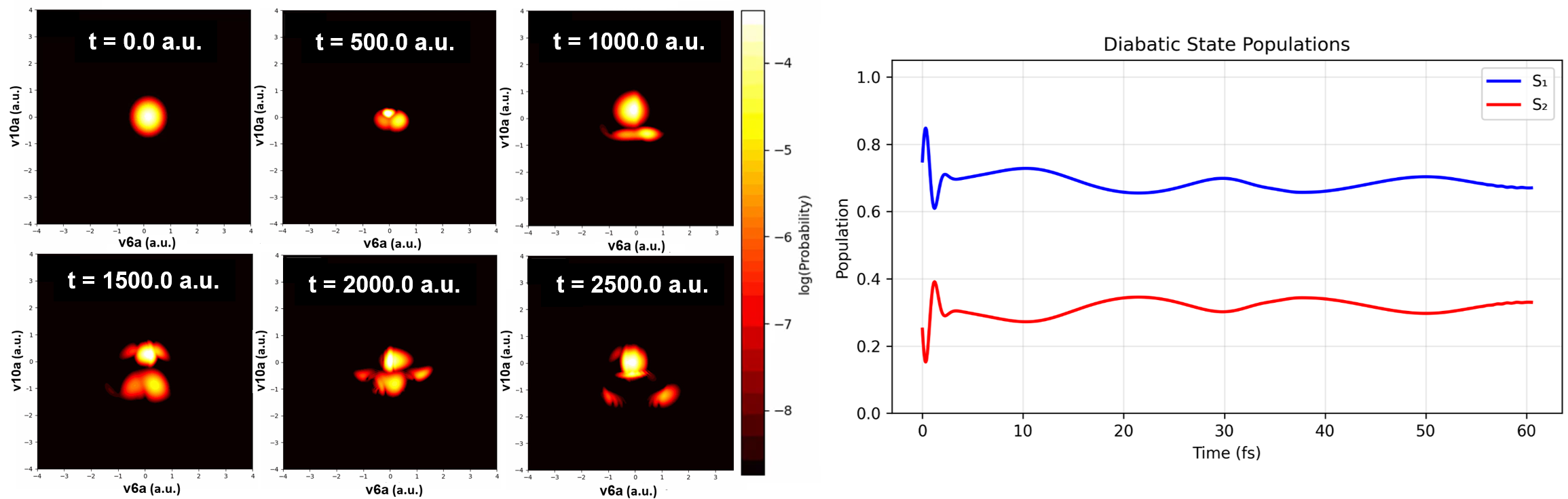
Figure 7: Population dynamics and probability heatmaps during wavepacket evolution on coupled pyrazine modes, showing recurrence and quantum scar phenomena.
Numerical results demonstrate robust recovery of known physical observables, correct Berry phase accumulation, and expected population transfer periods for both integrable and non-integrable regimes.
Resource Scaling and Fault-Tolerant Analysis
REQWIEM's explicit resource scaling is detailed for all operator classes:
- Rotations: Derived closed-form scaling, e.g., d8 for on-diagonal terms; d9 for off-diagonal.
- Circuit Depth: Depth scales with chromatic index N0, yielding N1 as a function of system symmetry.
- Ancilla Optimization: Fan-out requires N2 ancillas, but aggressive reuse and scheduling substantially minimize overhead.
REQWIEM is benchmarked directly against the QROM-based, phase-gradient approach of Motlagh et al. (May-Mann et al., 16 Mar 2026). Accounting for all system and precision corrections (grid size N3, phase register size N4, included bilinear terms, and realistic Trotter error budgets), REQWIEM achieves significant N5-depth and overall depth advantage—often exceeding an order of magnitude for N6 and N7. Parallelization via fan-out and chromatic scheduling provides pronounced depth savings, whereas serial QROM architectures are bottlenecked by shared phase accumulators.
Extensive tabulated resource comparisons are provided, e.g.,
| System |
Modes (N8) |
Channels (N9) |
Toffoli Layers (QROM) |
Rotation Layers (REQWIEM) |
Depth Ratio |
| Pyrazine |
24 |
2 |
Ω(t)0 |
Ω(t)1 |
Ω(t)2 |
| Ω(t)3-Anth |
19 |
5 |
Ω(t)4 |
Ω(t)5 |
Ω(t)6 |
| Full Anth/CΩ(t)7 |
246 |
4 |
Ω(t)8 |
Ω(t)9 |
n20 |
Implications and Theoretical Significance
The REQWIEM architecture provides several crucial implications for quantum simulation of chemistry and beyond:
- Extensibility and Generality: The circuit primitive—applying dense, position-dependent basis operators with explicit UCR and Clifford decompositions—is extendable beyond chemistry to any coupled-channel, grid-based quantum dynamics, including quantum materials, diffusion, and non-equilibrium statistical models.
- Parallelism as a Fundamental Lever: By maximizing physical and algorithmic parallelism (across modes, channels, and allowed operator classes), quantum simulation can amortize the exponential scaling required by the information content of the Hamiltonian. This demarcates a principal separation from oracle- or accumulator-based approaches.
- Practical Feasibility: Classical emulation confirms algorithmic behavior for system sizes at the edge of classical tractability (n21 per mode, n22), and resource analysis provides a clear roadmap for deployment on future fault-tolerant devices.
Prospects for Future Quantum Simulation
REQWIEM opens promising directions for both theoretical exploration and practical application:
Conclusion
REQWIEM constitutes a rigorous, explicit, oracle-free quantum algorithm for nonadiabatic quantum molecular dynamics, extending product-formula simulation into the regime of strongly coupled, high-dimensional, and multi-channel systems where oracle-based strategies are infeasible. Empirical and theoretical resource analysis demonstrates REQWIEM's strong advantage in n23-depth and practical circuit depth, attributed to principled exploitation of parallelism and structural decomposition. Beyond chemistry, the algorithm is directly extensible to a wide class of quantum simulation problems, particularly those structured as coupled, grid-based dynamical systems with nontrivial topologies or entanglement features. This work establishes a foundation and methodology by which quantum computational advantage in real-time simulation of complex, chaotic quantum systems may be realized as hardware scales.