- The paper presents a novel inference-time search framework, RosettaSearch, that leverages LLMs and structural feedback to iteratively optimize protein sequences.
- It demonstrates significant improvements in structural fidelity, with an 18.1% increase in pLDDT, 56.9% in TM-score, and design success rates improved up to 3.5×.
- The approach supports multi-objective optimization and multimodal feedback, offering a flexible, model-agnostic alternative to training-intensive methods.
Multi-Objective Inference-Time Sequence Search for Protein Design: An Analysis of RosettaSearch
Introduction and Motivation
Protein sequence design conditioned on a specific backbone is a critical bottleneck in computational biology, with direct implications for experimental viability in enzyme engineering, therapeutics, and de novo biomolecule design. Current state-of-the-art models such as LigandMPNN and ProteinMPNN, optimized primarily for sequence recovery by single-pass autoregressive decoding, frequently fail to guarantee that the predicted sequence will reliably fold into the desired target structure when evaluated by an external structural oracle. This limitation arises from the inability of these models to revise or self-correct their outputs in response to structural feedback at inference time. Consequently, a large fraction of generated sequences are predicted with low confidence or depart geometrically from the intended backbone—an issue that incurs substantial downstream experimental cost.
RosettaSearch (2604.17175) proposes an inference-time, multi-objective search framework that leverages LLMs as generative optimizers, orchestrated via a priority-based search algorithm and guided by feedback from structure prediction models (notably, RosettaFold3). The method aims to bridge the gap between computational sequence design and experimental reliability by enabling iterative, feedback-driven refinement of candidate sequences while allowing for rapid adaptation to diverse objectives without model retraining.
Algorithmic Framework
RosettaSearch operates by coupling LLMs (and optionally VLMs) to a black-box structure prediction oracle. Rather than one-shot sequence generation, the LLM is tasked with proposing targeted edits to a candidate sequence in response to structured feedback, which combines both global scalar rewards and fine-grained, residue-level annotations.
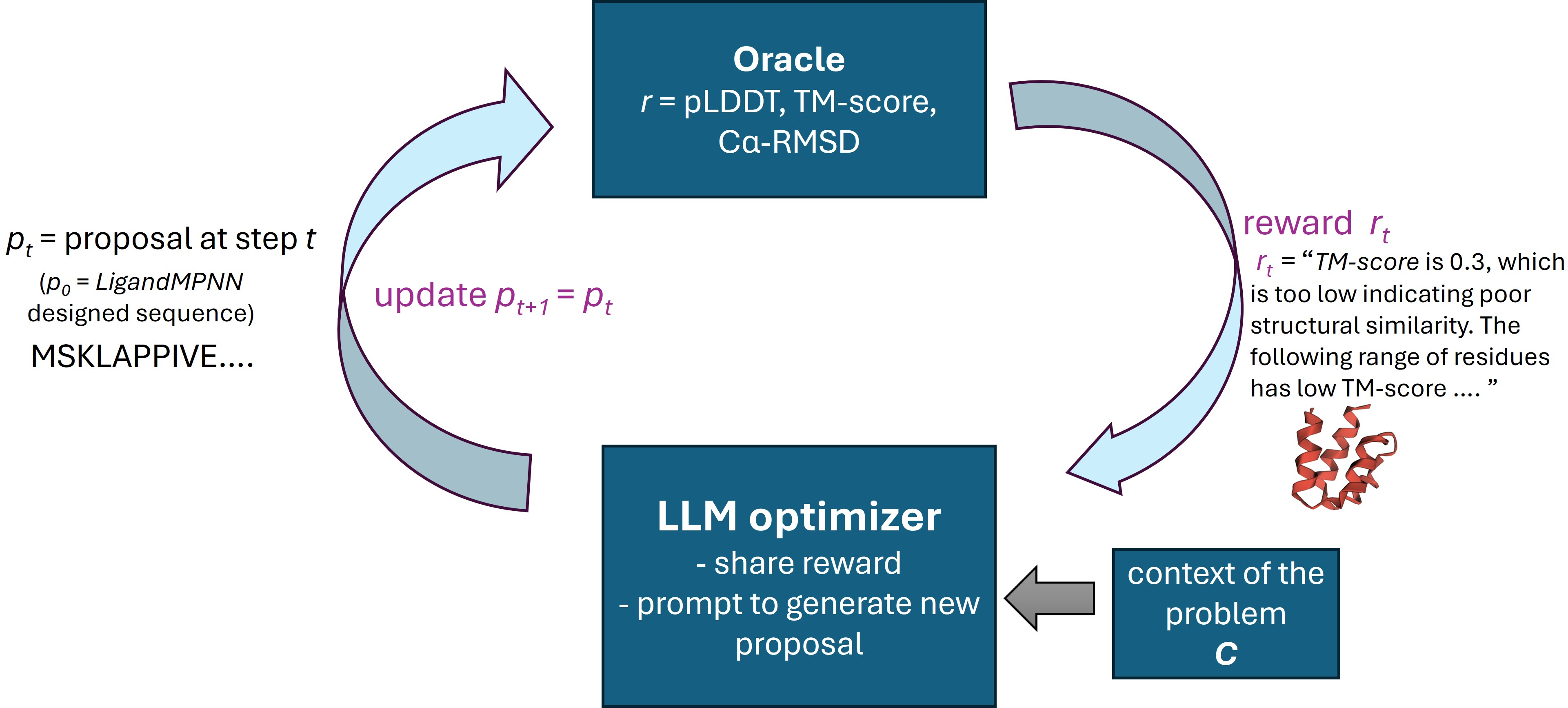
Figure 1: Schematic of the RosettaSearch approach for inference-time, feedback-driven protein sequence refinement.
Two major iterative optimization schemes are examined:
- Sequential Revision: An LLM proposes single modifications in a greedy, autoregressive fashion. At each iteration, feedback from RosettaFold3 is provided, and the optimizer proposes edits accordingly. However, this approach is susceptible to premature convergence and irrecoverable error accumulation.
- Priority Search: A broader, priority-based parallel search that maintains a buffer of top-K candidates at each iteration, generating and evaluating multiple candidate modifications concurrently. This approach enables better exploration, mitigates local minima, and is empirically shown to outperform sequential strategies.
RosettaSearch supports multi-objective optimization, where rewards for structural fidelity (pLDDT, TM-score, Cα-RMSD) are combined into a single scalar with tunable weights. Crucially, local textual feedback (identifying problematic structural regions) is found to be essential for effective optimization, as global scalar signals alone are insufficient to drive meaningful refinement.
A further extension incorporates multimodal feedback: by using VLMs, the optimizer ingests both structured text and rendered images of predicted protein structures, providing spatial context that can enrich the LLM's reasoning trace.
Experimental Evaluation
RosettaSearch is evaluated on multiple backbone-conditioned design scenarios:
- PDB-derived monomer dataset (400 proteins), using both native sequences and LigandMPNN outputs as starting points.
- The Dayhoff dataset, featuring de novo RFDiffusion backbones with ProteinMPNN-designed sequences, constituting a challenging, high-fidelity testbed.
- A set of optimal binders from the BindCraft study, designed via computationally intensive pipelines.
- Random sequence initialization for benchmarking limits of optimization.
Structural fidelity is measured by per-sequence pLDDT, TM-score, and Cα-RMSD, with design success rates defined as the fraction of sequences achieving pLDDT≥0.8 and TM-score≥0.8. Stringent multi-objective criteria further consider Cα-RMSD ≤1.5.
Notable findings:
- Structural Accuracy Improvements: For LigandMPNN-initialized sequences, RosettaSearch yields an 18.1% improvement in pLDDT, 56.9% in TM-score, and a 31.2% reduction in CA-RMSD, with multi-objective optimization providing even greater gains.
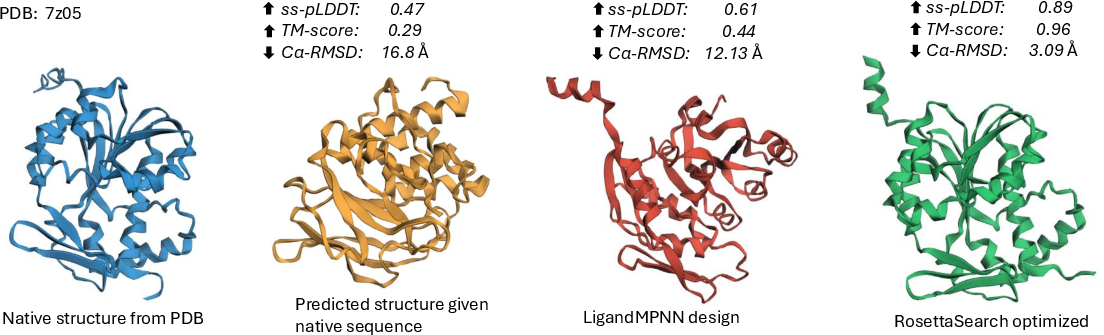
Figure 2: Benchmark example of inference-time RosettaSearch optimization; starting from a low-fidelity LigandMPNN sequence, the final design approaches native structural metrics.
- Design Success Rate: Starting from LigandMPNN sequences, RosettaSearch increases the design success rate from 7.9% to 20.5%, a 2.5× improvement under RosettaFold3 evaluation. Stringent criteria (all three objectives) yield a 3.5× increase in the number of successful designs relative to the baseline.
- Generalization: When evaluated with an independent structure oracle (Chai-1), gains are preserved—the success rate increases from 3.1% to 6.7%, demonstrating resistance to overfitting model-specific inductive biases.
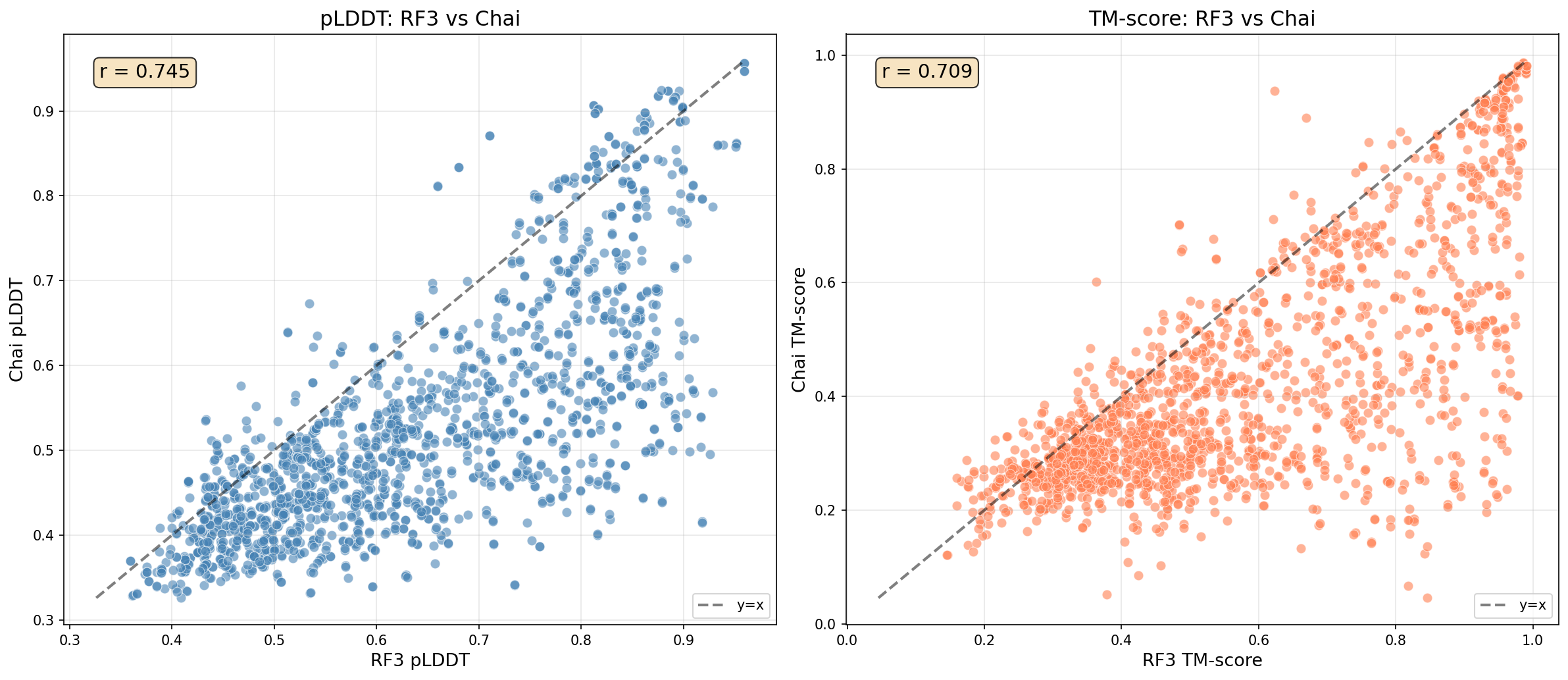
Figure 3: Cross-model evaluation of structural scores for designs optimized by RosettaSearch, showing improvements on both RF3 and Chai-1 predictors.
- Model Family Independence and Scaling: Comparable success rates (19.4% for Gemini-3, 20.5% for o4-mini) across LLM families, with improvements scaling with reasoning ability.
- BindCraft Binders: RosettaSearch improves already optimized binders—raising pLDDT from 0.85 to 0.90 and reducing CA-RMSD from 1.37 Å to 0.99 Å—demonstrating utility for post hoc refinement.
- Dayhoff Evaluation: On the high-fidelity Dayhoff set, success rate rises from 72.4% (ProteinMPNN) to 89.5% (RosettaSearch), and stringently from 45.5% to 67.3%.
- Optimization Trajectory: Consistent monotonic improvements in metrics are observed throughout optimization.
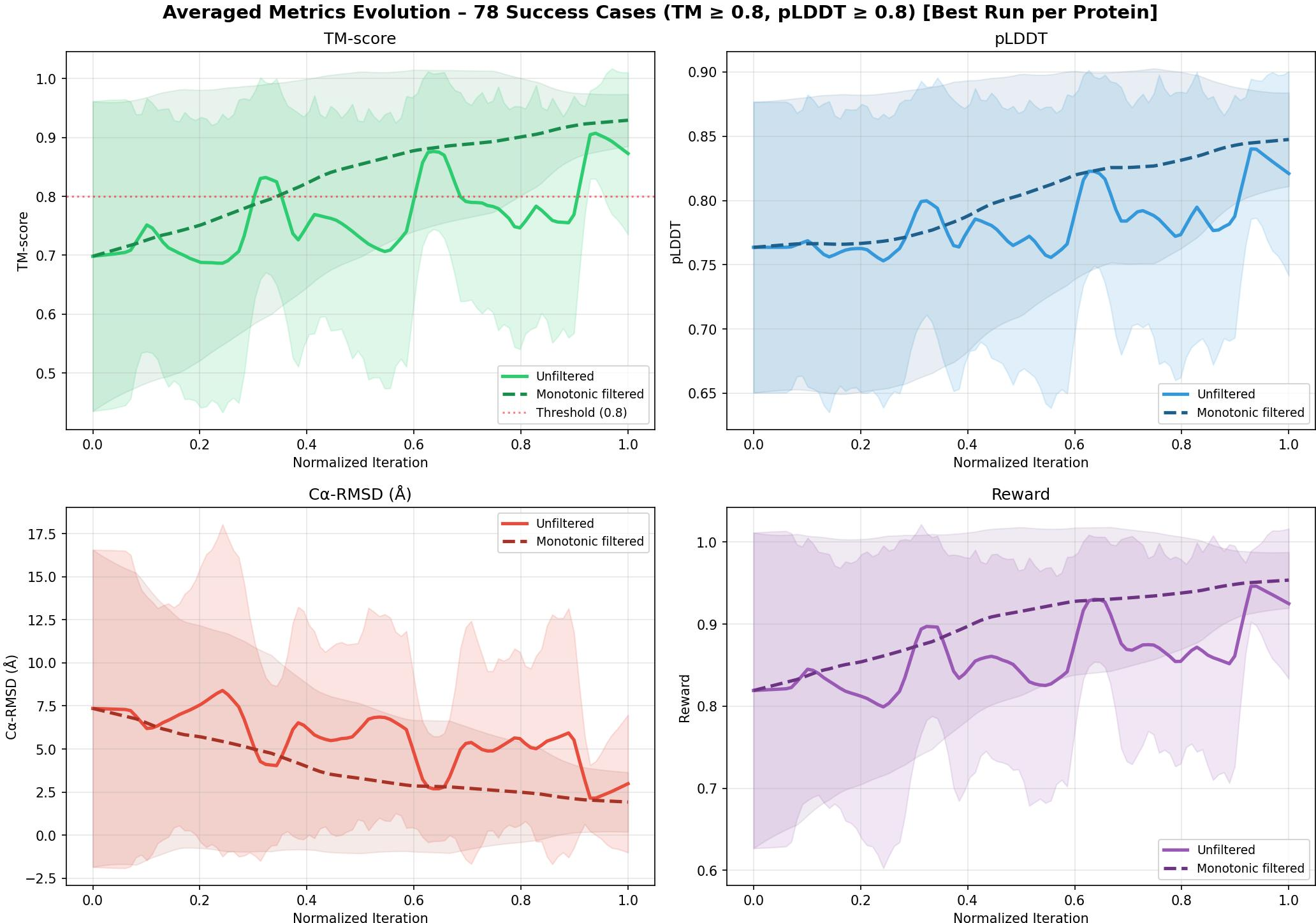
Figure 4: Metric and reward evolution during optimization, demonstrating steady improvements across successful redesigns.
Sequence Diversity and Edit Patterns
Analysis of generated sequence diversity (Figure 5) reveals a broad distribution of sequence identity changes, with a mean identity of 82.4% but a long tail of more aggressively redesigned sequences. Additionally, position-wise analyses (Figure 6) indicate that the optimizer targets problematic residues highlighted by local feedback, but can also propose global redesigns when necessary.
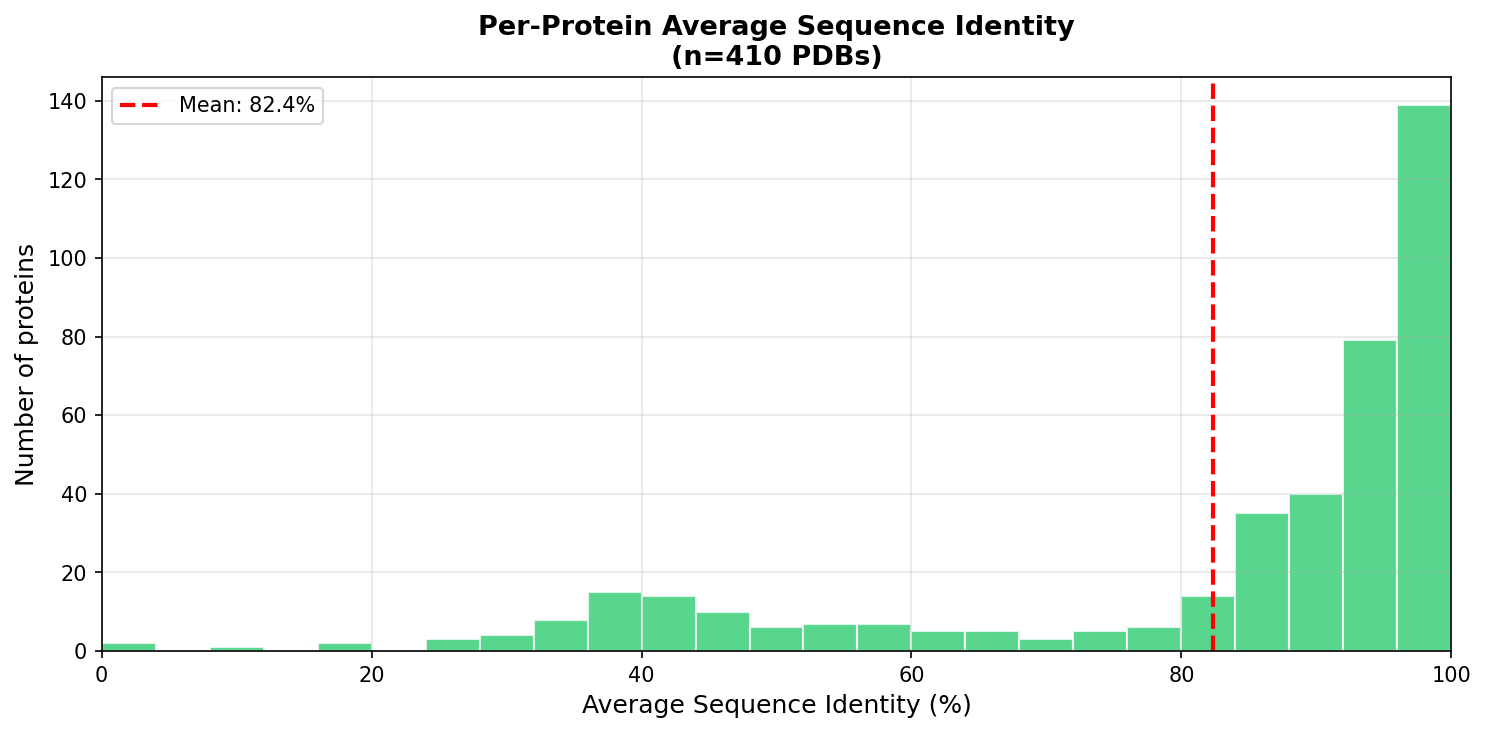
Figure 5: Distribution of sequence identity between starting and optimized sequences, reflecting adaptive edit magnitudes.
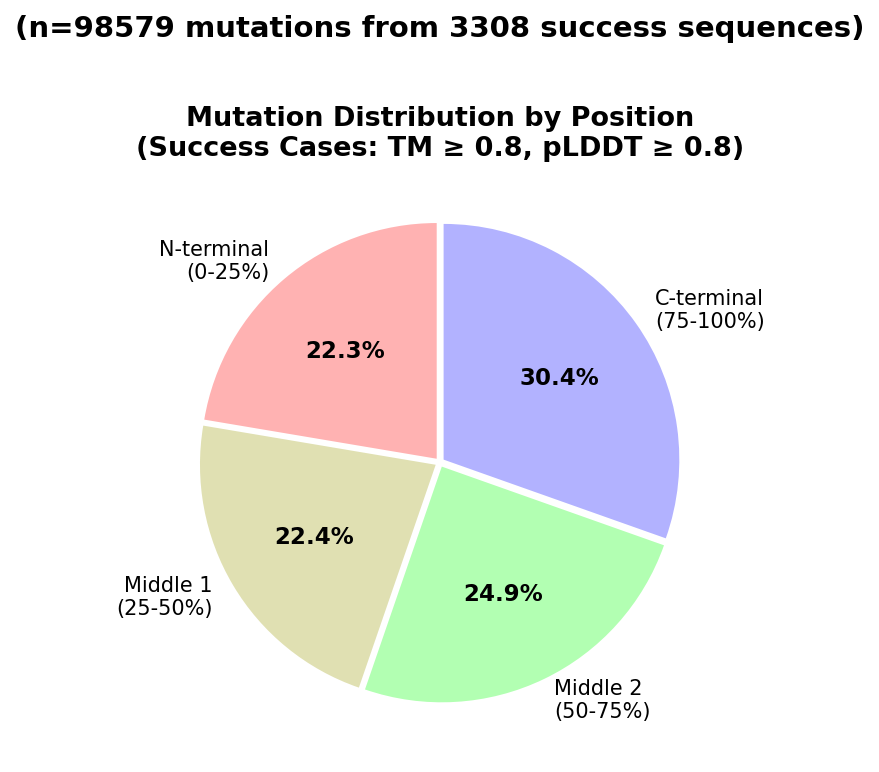
Figure 6: Histogram of residue positions modified during optimization among successful designs.
Expert Evaluation and Multimodal Reasoning
Samples of reasoning traces (Figure 7) were annotated by biochemistry experts and by LLMs acting as reviewers. While many reasoning steps were consistent and chemically plausible, domain experts noted that some sequence edits, though locally sensible, could be non-optimal in the context of specific backbone features—highlighting the inherent limitation of LLM-only reasoning and the importance of rigorous structural feedback.

Figure 7: Example of expert annotation and LLM-generated reasoning for a single sequence optimization step.
VLM-based multimodal optimization yields richer reasoning traces and incorporates structural spatial knowledge, but numerical gains are comparable to text-only modes under current algorithms.
Practical and Theoretical Implications
RosettaSearch establishes that inference-time optimization, even with a fixed generative model, can substantially improve protein sequence design fidelity. This result demonstrates that controlled inference-time search—backed by LLM-driven reasoning and robust oracular feedback—can outperform single-pass generation, and in certain regimes compete with or supersede approaches reliant on retraining, RL, or preference-fine-tuning.
The framework is agnostic to both LLM family and structure oracle, readily adapts to new objectives, and enables search-based fine-tuning without labeled training data. The post-processing trajectories generated during optimization are valuable for model post-training or as augmentation data, facilitating improved generalization.
Limitations include a focus on single-sequence fidelity rather than explicit diversity objectives, and susceptibility to feedback misalignment or reward hacking in the absence of well-calibrated local feedback.
Future Prospects
- Extension to explicitly diversity-aware sequence search, to generate panels of functionally diverse, high-fidelity candidates.
- Tighter integration of domain constraints using richer multimodal feedback beyond static structure images, potentially via molecular simulation traces.
- Combination with experimental-adaptive design loops, closing the gap between in silico optimization and wetlab validation.
- Application of analogous inference-time generative optimization approaches to other discrete scientific design problems.
Conclusion
RosettaSearch demonstrates that LLMs, deployed as generative optimizers within a structured, feedback-driven, inference-time search framework, can substantially improve the fidelity and reliability of protein sequence design. The empirical gains in structural accuracy, generalization across datasets and evaluators, and robustness to sequence initialization establish the paradigm as a powerful, flexible alternative to training-intensive methods. This work motivates further development of reasoning-driven inference-time optimization strategies for complex, high-dimensional scientific design challenges.

