- The paper introduces a novel sequence-centric stochastic beam search that leverages MLM pseudo-log-likelihood for scalable protein engineering.
- The paper demonstrates that SBS beam search outperforms traditional mutation-centric methods in generating diverse, high-probability antibody sequences.
- The paper shows that integrating multi-objective guidance, including auxiliary predictors, achieves up to 100% experimental success in synthesizability and binding.
Optimizing Protein Engineering with Masked LLMs: Sampling Methods and Multi-Objective Guidance
Introduction
The application of large masked LLMs (MLMs) to antibody and general protein engineering has become a major area of interest due to the combinatorial complexity and cost of experimental sequence optimization. This paper ("How to make the most of your masked LLM for protein engineering" (2603.10302)) systematically investigates the efficacy of sequence generation algorithms in this context. The focus is on leveraging protein MLMs for iterative improvement of antibody therapeutics via sequence proposal, especially addressing the gap in principled sampling methods and scalable integration of auxiliary objectives.
The Problem Setting: Protein Engineering with MLMs
Protein and antibody optimization campaigns use iterative machine learning-guided design cycles. At each iteration, a chosen set of high-performing candidate sequences is used as seeds, and a generative model proposes new variants. Optionally, auxiliary predictive models (for properties such as binding or manufacturability) may be incorporated to bias the generation process. The resulting sequences are experimentally validated, and the cycle repeats.
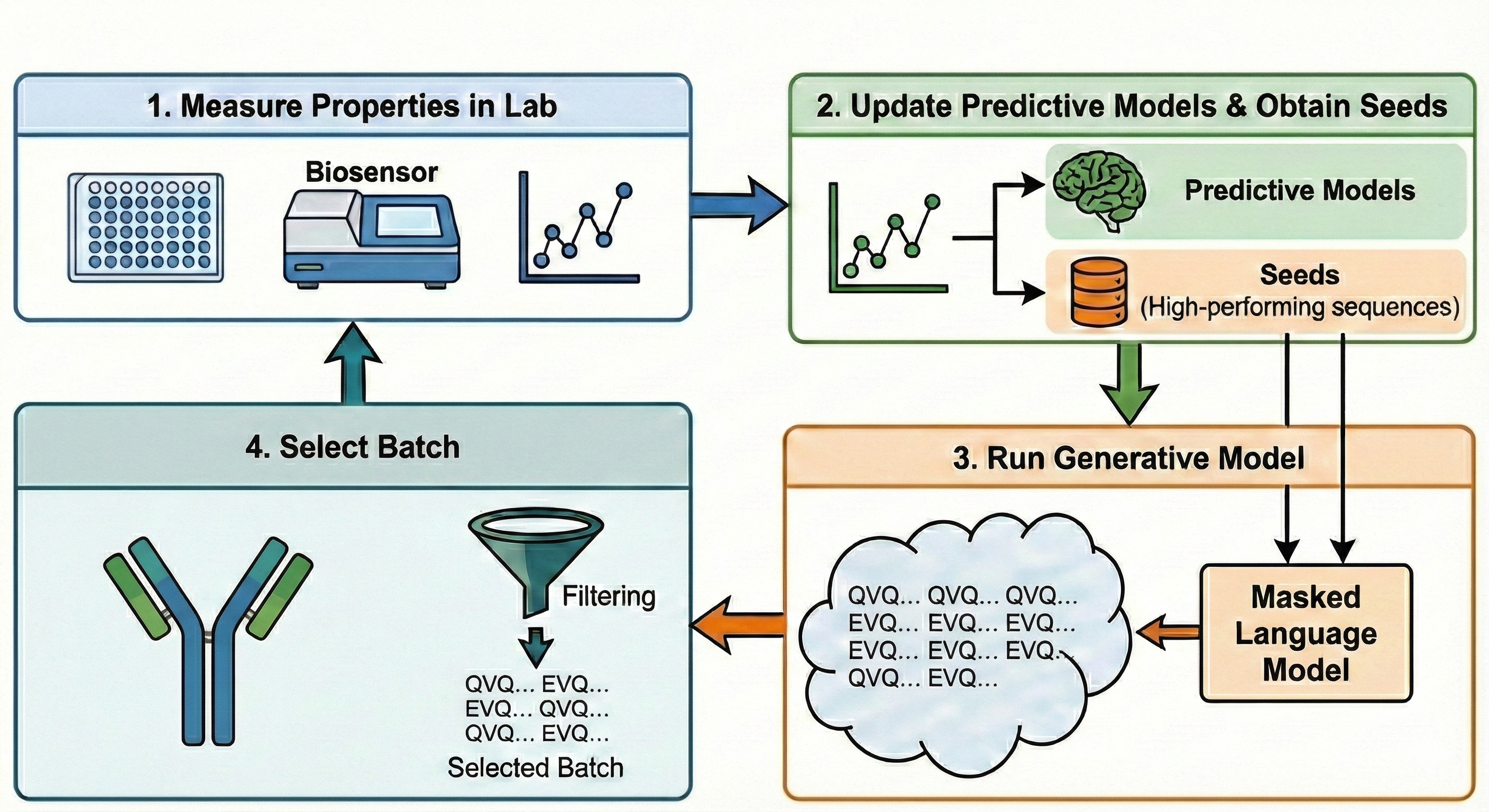
Figure 1: Machine learning-guided iterative design of therapeutic antibodies; this work focuses on Step 3, sequence proposal via MLM-guided sampling and guidance.
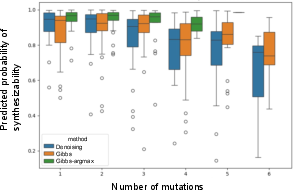
While the development of high-performing protein MLMs (e.g., ESM-2, Sapiens, AbLang2) has rapidly expanded, there has been little rigorous analysis of how the choice of sampling/generation algorithm influences the quality and diversity of designed sequences. Traditional mutation-centric sampling methods, adapted from denoising and Gibbs sampling for text, are not computationally scalable and are prone to generating low-likelihood, non-functional sequences. Mutation-based samplers also struggle to flexibly integrate additional objectives, particularly when the auxiliary scores are non-differentiable or require complete sequences.
Proposed Approach: Sequence-Centric Stochastic Beam Search
The authors introduce a sequence-centric sampling framework based on temperature-annealed stochastic beam search (SBS) guided by the MLM's pseudo-log-likelihood (PLL). They exploit the fact that for a given sequence, the MLM can efficiently provide approximate PLLs for all possible single-residue mutants, leveraging the wild-type marginal approximation. By iteratively expanding the beam across a fixed number of allowed substitutions, the method enables the rapid and diverse exploration of local sequence neighborhoods.
Key features and advantages:
- Computational Efficiency: The cost per sequence is reduced by orders of magnitude (factor ∼20EL) compared to classical mutation-centric methods for realistic parameters.
- Quality of Proposals: The method generates sequences with higher MLM PLL (implying higher model confidence, often correlating with functionality).
- Flexibility: Diverse batches are attainable by controlling SBS temperature and Gumbel perturbation, independent of proposal uniqueness.
Integrating Multi-Objective Guidance
In realistic protein design, the MLM likelihood should be balanced with explicit optimization for other molecular properties (e.g., synthesizability, binding, developability). The framework is agnostic to the nature of auxiliary objectives; scalarizations such as Pareto non-dominated sorting and smooth Tchebycheff scalarization (STS) can be directly applied.
This black-box multi-objective optimization (MOO) capability enables guidance using:
- Differentiable and non-differentiable sequence functions
- Arbitrary property predictors trained on limited in vitro data
- Scalarization strategies tunable for exploration vs. exploitation
Experimental Evaluation: In Silico and In Vitro
In Silico Benchmarking
The study benchmarks multiple protein MLMs (ESM-2, AbLang2, AMPLIFY, Sapiens) and CLMs (pIgGen, CloneLM) across a large set of antibody sequence design tasks. Sampling algorithms evaluated include denoising, Gibbs, Gibbs-argmax, and beam search (proposed). MLMs are evaluated for their ability to generate sequences with target numbers of substitutions, as well as their impact on metrics: predicted synthesizability, thermostability, mutational diversity, humanness, isoelectric point, and CDR targeting.
Key findings:
In Vitro Head-to-Head Antibody Optimization
A real-world antibody engineering campaign was conducted, with candidate designs proposed by different model/sampler pairs and experimentally validated for synthesizability, binding, and developability-related features. The experimental protocol included both unsupervised sequence generation and various forms of supervised filtering, ranking, and MOO-guided sequence selection leveraging property predictors trained on initial data.
Numerical results and claims:
- Unsurprisingly, ESM2-650M and AbLang2 achieved the highest rates of generating experimentally successful binders without auxiliary guidance.
- SBS beam search produced higher success rates and tighter output distributions than mutation-centric algorithms (e.g., Gibbs sampling) across matched models.
- Integration of supervised guidance (STS scalarization) led to a 100% experimental success rate in the context of synthesizability and binding, outperforming both unsupervised and post-hoc filtered results.
- However, strong guidance increased risks such as decreased humanness and elevated bias toward specific sequence motifs, underscoring the necessity for judicious selection of objectives and weights in MOO.
Implications and Future Directions
This paper establishes that the methodological gap in sampling from protein MLMs can have an impact comparable to that of model selection in antibody engineering campaigns. The introduced framework, SBS via the wild-type marginal approximation, enables practical, scalable, and high-quality design, supporting the rapid exploration of local and global mutant neighborhoods.
The demonstration that multi-objective, supervised guidance can drive success rates to 100% (albeit with potential for overfitting to supervised signals and reduction in desired diversity or humanness) has two main implications:
- Practical: High-throughput protein design can be efficiently achieved with smaller, domain-specific MLMs when paired with effective sampling and guided optimization strategies—lowering experimental costs and resource use.
- Theoretical: Flexible, black-box guidance for sequence generation expands the scope of ML-guided protein engineering, but introduces new trade-offs in sequence diversity and multi-property balancing.
Further research will be needed to:
- Develop adaptive, diversity-promoting sampling strategies that balance exploitation of model likelihood and robustness to overfitting via auxiliary predictors.
- Extend this framework to broader classes of proteins and multi-domain sequences.
- Automate and formalize the selection of objective weights or design constraints for diverse therapeutic goals.
Conclusion
This work delineates the critical importance of sequence-centric, beam-based sampling and black-box, multi-objective guidance for effective protein MLM-driven antibody engineering. The approach efficiently leverages MLMs for local sequence space exploration, outperforms established mutation-centric methods, and generalizes to guided design with auxiliary objectives. Its practical recommendations—utilize effective sampling, combine with explicit supervision, and prefer sequence-centric search—set a roadmap for advancing data- and compute-efficient protein engineering across therapeutic modalities.