- The paper presents a hybrid framework that combines low-cost QM evaluations with fragment-based GNN corrections for scalable and chemically accurate PES prediction.
- It employs state-of-the-art architectures (MXMNet and PAMNet) with a multi-stage curriculum training strategy to address the imbalance in energy distributions of complex molecular clusters.
- The study demonstrates effective data-adaptive transfer learning via teacher–student knowledge distillation, enhancing model generalization to unseen molecular configurations.
Transferable FB-GNN-MBE: Data-Adaptive Transfer Learning in Deep Learned Many-Body Expansion Theory
Introduction
Accurate modeling of potential energy surfaces (PES) for large molecular assemblies is a fundamental challenge in computational chemistry, with direct implications for understanding solution-phase phenomena, biomolecular conformations, and catalysis. Traditional quantum mechanical (QM) approaches—such as MP2 or DFT—are computationally prohibitive for systems with hundreds of atoms. While classical force fields offer expediency, they lack fidelity in describing quantum-chemical phenomena, including non-covalent effects and charge fluctuations. The many-body expansion (MBE) decomposes total energies into sums of interactions between fragments, but direct QM evaluation still incurs prohibitive costs at high n-body orders.
Recent advances in machine learning, including neural networks (NNs) and graph neural networks (GNNs), have provided efficient surrogates for PES prediction. However, standard GNNs typically ignore chemical hierarchy and fragment distinctions, limiting both transferability and physical interpretability, particularly in non-covalent, hierarchically assembled systems. The present work introduces FB-GNN-MBE, a framework that integrates fragment-based GNNs (FB-GNNs) into the MBE formalism, enabling accurate, scalable prediction of PESs for complex systems via data-adaptive transfer learning and knowledge distillation.
FB-GNN-MBE Framework: Architecture and Methodology
Many-Body Expansion with Fragment-Based GNNs
FB-GNN-MBE decomposes the total energy of an N-fragment system as
E≈i∑NEi1B+i<j∑NEij2B+i<j<k∑NEijk3B,
with 1B energies evaluated directly using low-cost QM (MP2 or DFT), and higher-order corrections (2B and 3B) predicted by trained FB-GNNs from instantaneous geometric configurations. This hybrid approach maintains chemical interpretability and drastically reduces computational overhead compared to traditional ab initio MBE calculations.
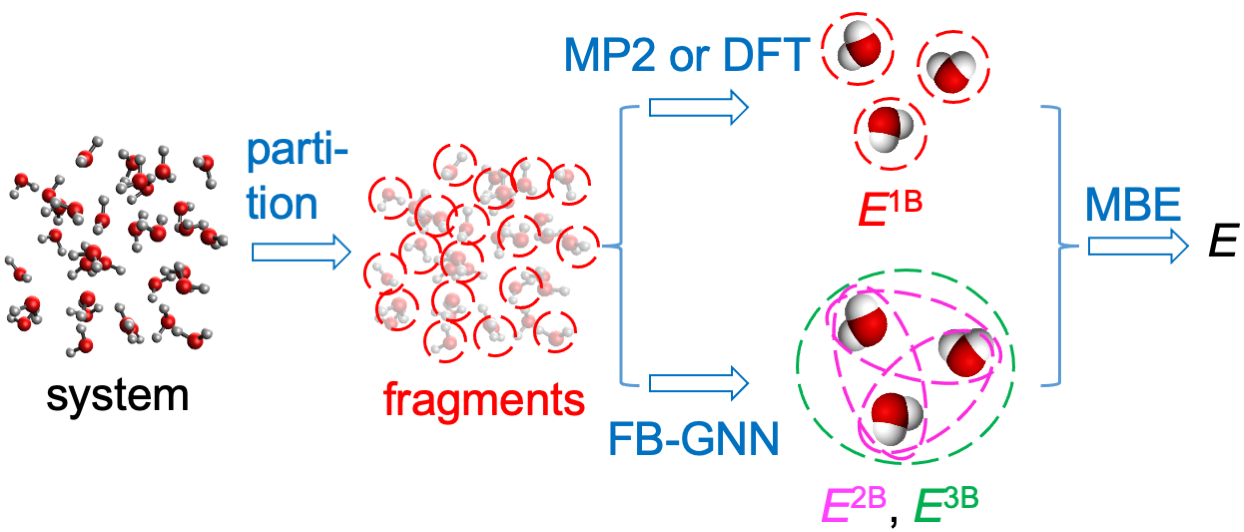
Figure 1: Schematic strategy of FB-GNN-MBE—1B energies via MP2/DFT, 2B and 3B corrections from FB-GNN-trained structure–property relationships.
FB-GNN Models: MXMNet and PAMNet
FB-GNN-MBE employs two state-of-the-art fragment-based GNN architectures: MXMNet and PAMNet. Both leverage multiplex hierarchical representations, modeling the system as a supergraph of interacting fragments (local graphs within a global graph), and utilize message-passing mechanisms to fuse local (intrafragment) and global (interfragment) information.
- MXMNet applies sequential global–local message passing with cross-layer mapping, updating both node embeddings and fragment representations iteratively.
- PAMNet operates parallel message-passing over global and local subgraphs, integrating outputs via attention pooling for greater representational flexibility and interpretability.
Physics-inspired embedding via radial and spherical basis functions, and E(3)-invariant features, ensures robust encoding of chemical environments and geometric dependencies.
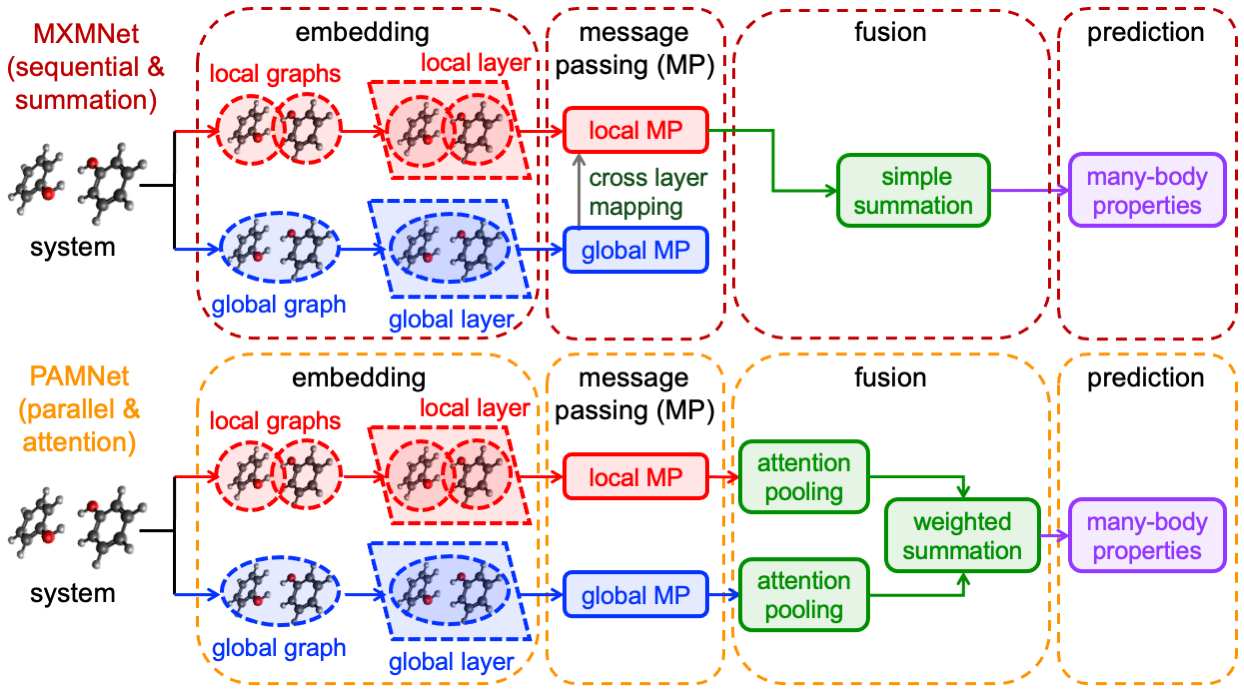
Figure 2: Schematic of MXMNet (top) and PAMNet (bottom) architectures for hierarchical chemical systems.
Robust Training on Complex Distributions
A challenge in fitting PES data arises from class imbalance, especially in mixed-density clusters where low-magnitude n-body energies dominate. FB-GNN-MBE addresses this via a multi-stage (curriculum) training strategy, progressively introducing data from high-magnitude to full energy range, ensuring effective learning of repulsive and near-equilibrium regions before refining long-range weak interactions.
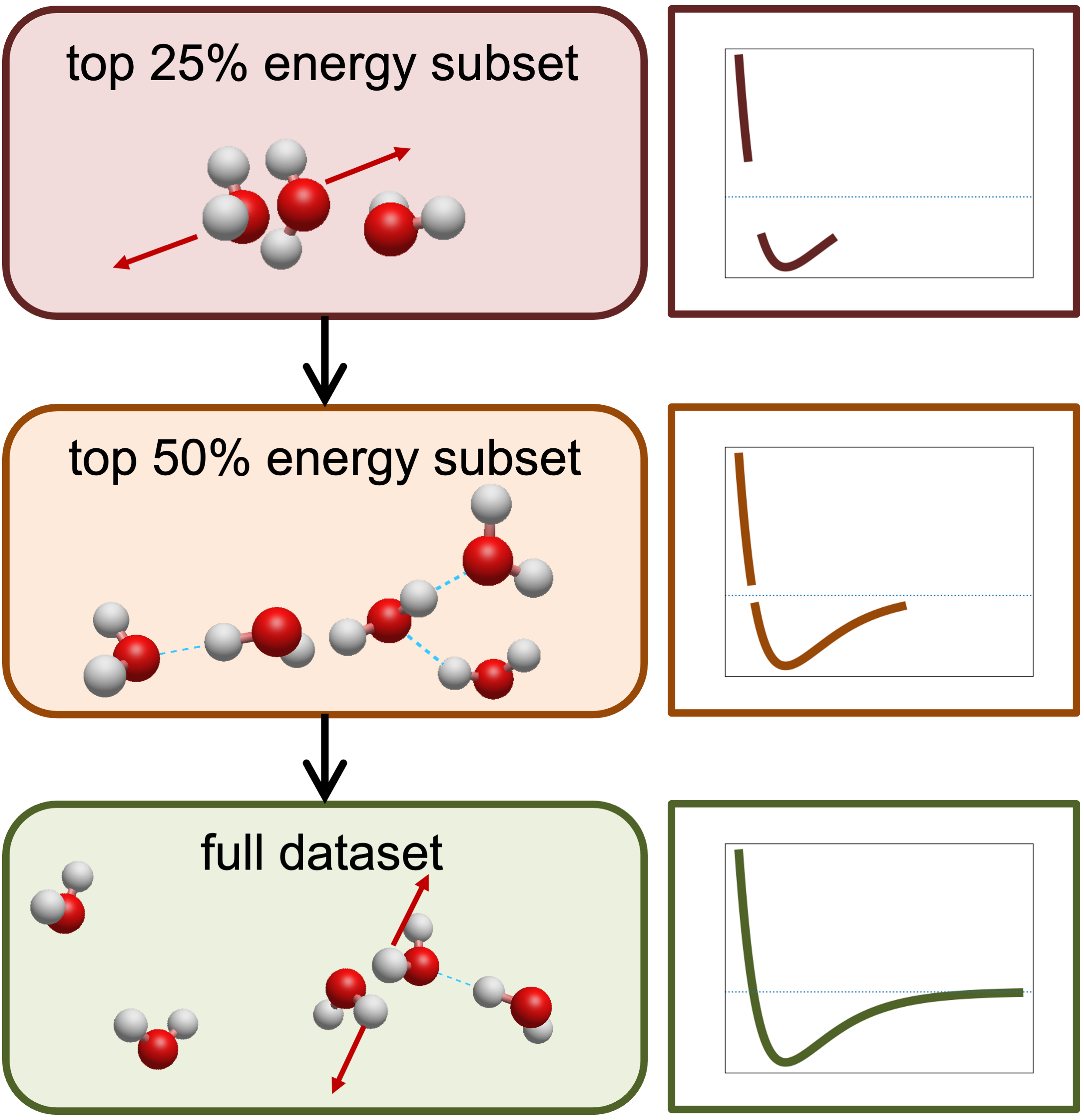
Figure 3: Multi-stage training strategy—progressive learning from high-energy to full PES landscape.
Transfer Learning via Teacher–Student Knowledge Distillation
To enable data-efficient adaptation to out-of-distribution or undersampled configurations, FB-GNN-MBE introduces a teacher–student knowledge distillation protocol. A heavy-weight teacher model (e.g., PAMNet trained on mixed-density clusters) supervises light-weight student GNNs (e.g., DimeNet, ViSNet, SchNet) via distillation over a reduced dataset. The process involves matching both energies and intermediate feature representations, followed by fine-tuning on minimal system-specific data. This mitigates overfitting and catastrophic forgetting, especially in small clusters or novel environments.
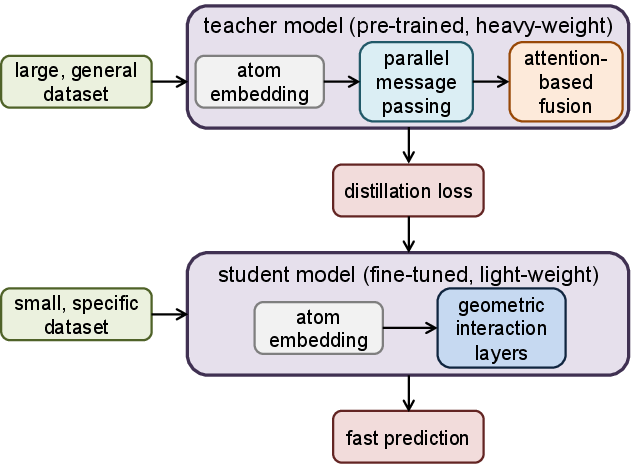
Figure 4: Teacher–student knowledge distillation—teacher (PAMNet) supervises student GNNs via soft energy and feature targets.
Overall Accuracy and Efficiency
FB-GNN-MBE demonstrates chemical accuracy in 2B and 3B energies across double-density water, phenol, and mixed clusters, achieving high R2 and sub-chemical threshold mean absolute errors (MAEs), while reducing computational cost by up to four orders of magnitude compared to MP2/DFT MBE.
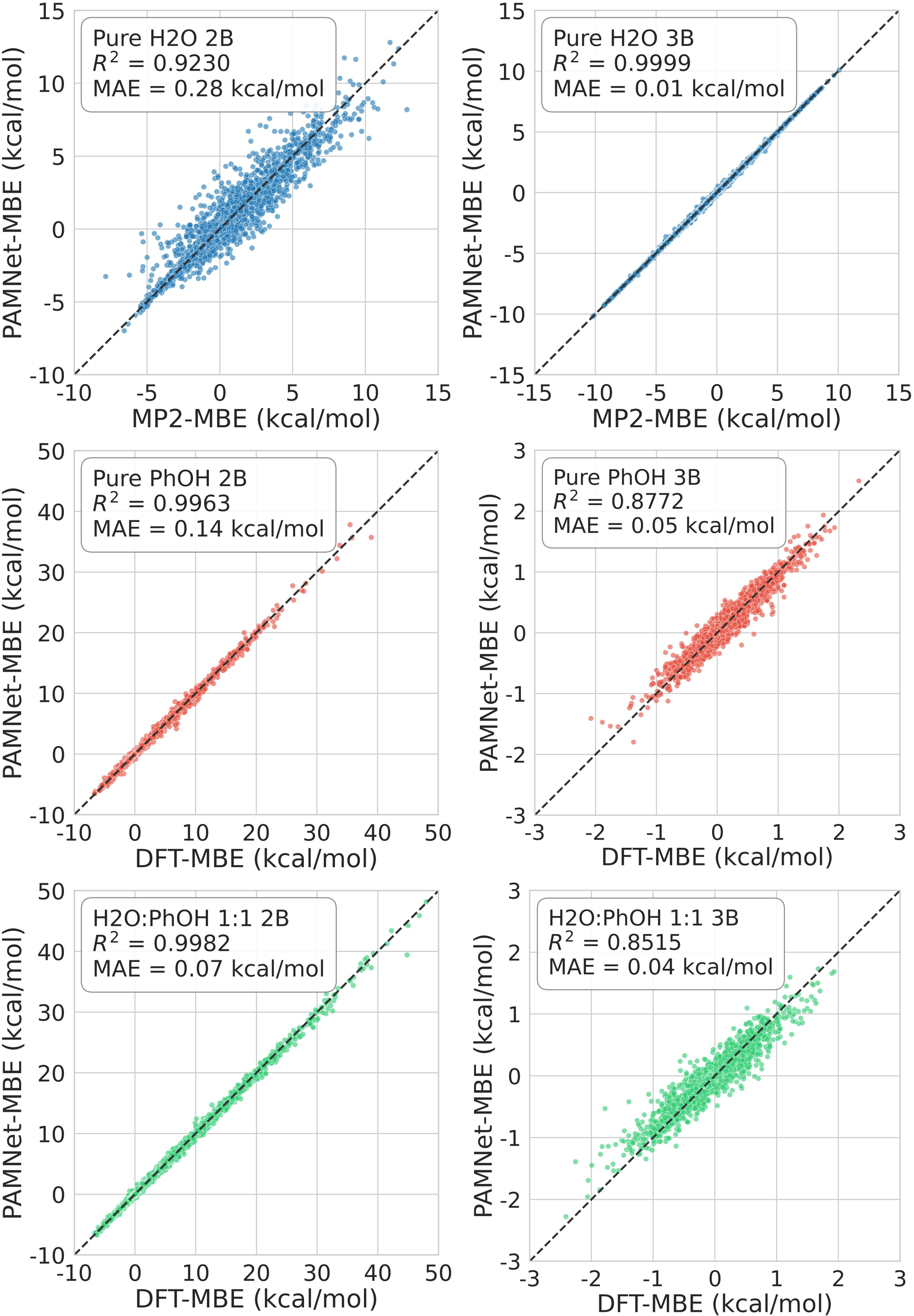
Figure 5: 2B (left) and 3B (right) energies on double-density water, phenol, and 1:1 water:phenol clusters as predicted by PAMNet-MBE (blue) versus MP2/DFT-MBE (red, reference).
Superiority Over Non-Fragment GNNs
Direct comparison shows that FB-GNN-MBE (both MXMNet-MBE and PAMNet-MBE) outperforms non-fragment GNN-MBE variants (SchNet, DimeNet, ViSNet, MACE, FragGen) in both accuracy and transferability, especially for 2B corrections where explicit fragment hierarchy is critical. Non-FB-GNNs perform relatively better only in 3B energies, reflecting their limitations in encoding long-range, chemically context-dependent interactions.
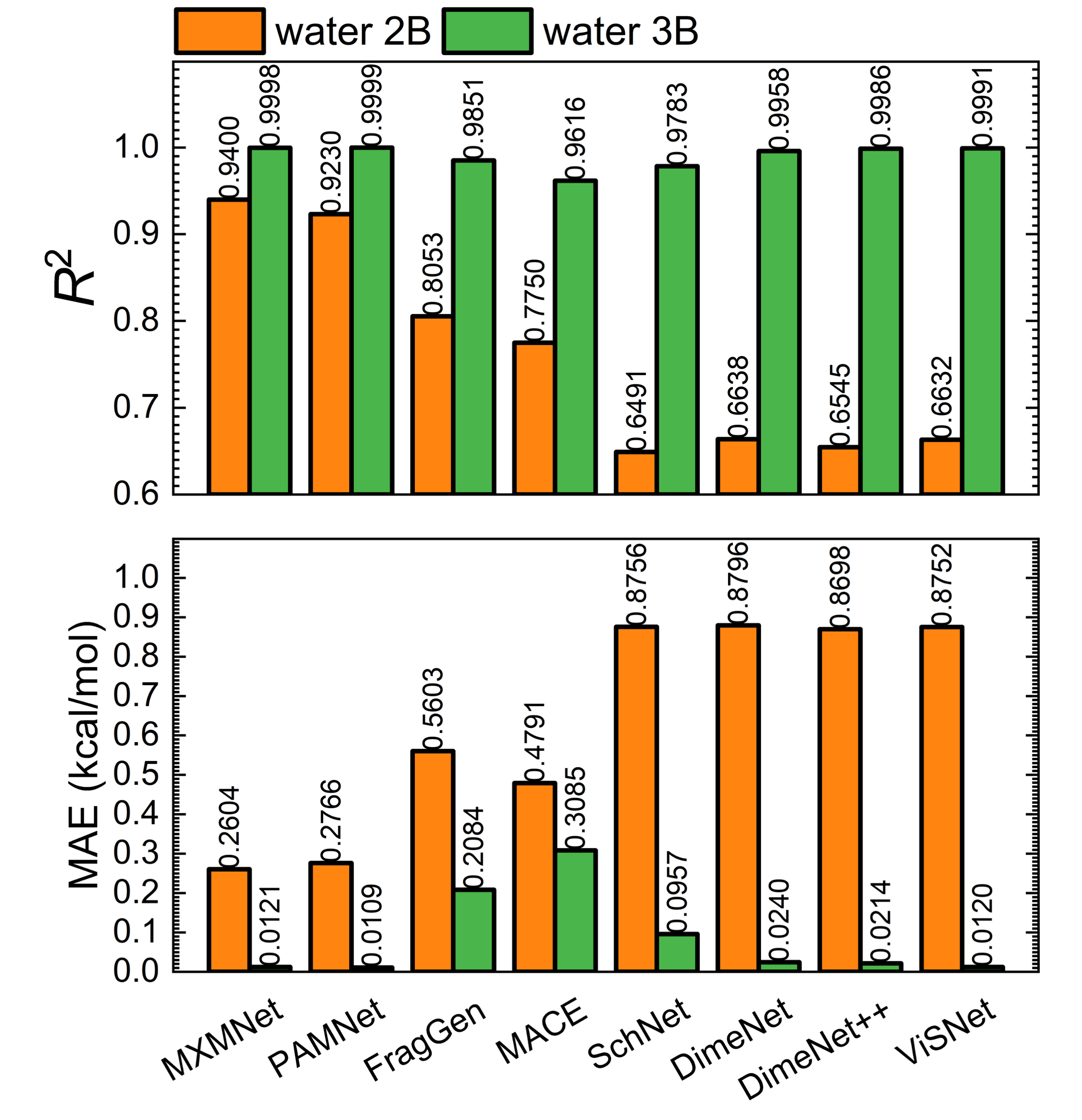
Figure 6: Performance metrics (R2 and MAE) for 2B and 3B energies: comparison between FB-GNN-MBE and non-FB-GNN-MBE on double-density (H2O)67.
Effectiveness of Multi-Stage Training
The multi-stage curricular approach resolves the imbalance in mixed-density datasets, restoring high accuracy for both 2B and 3B energies. The protocol allows the model to capture both high-energy and near-dissociation configurations, circumventing bias toward trivial zero predictions.
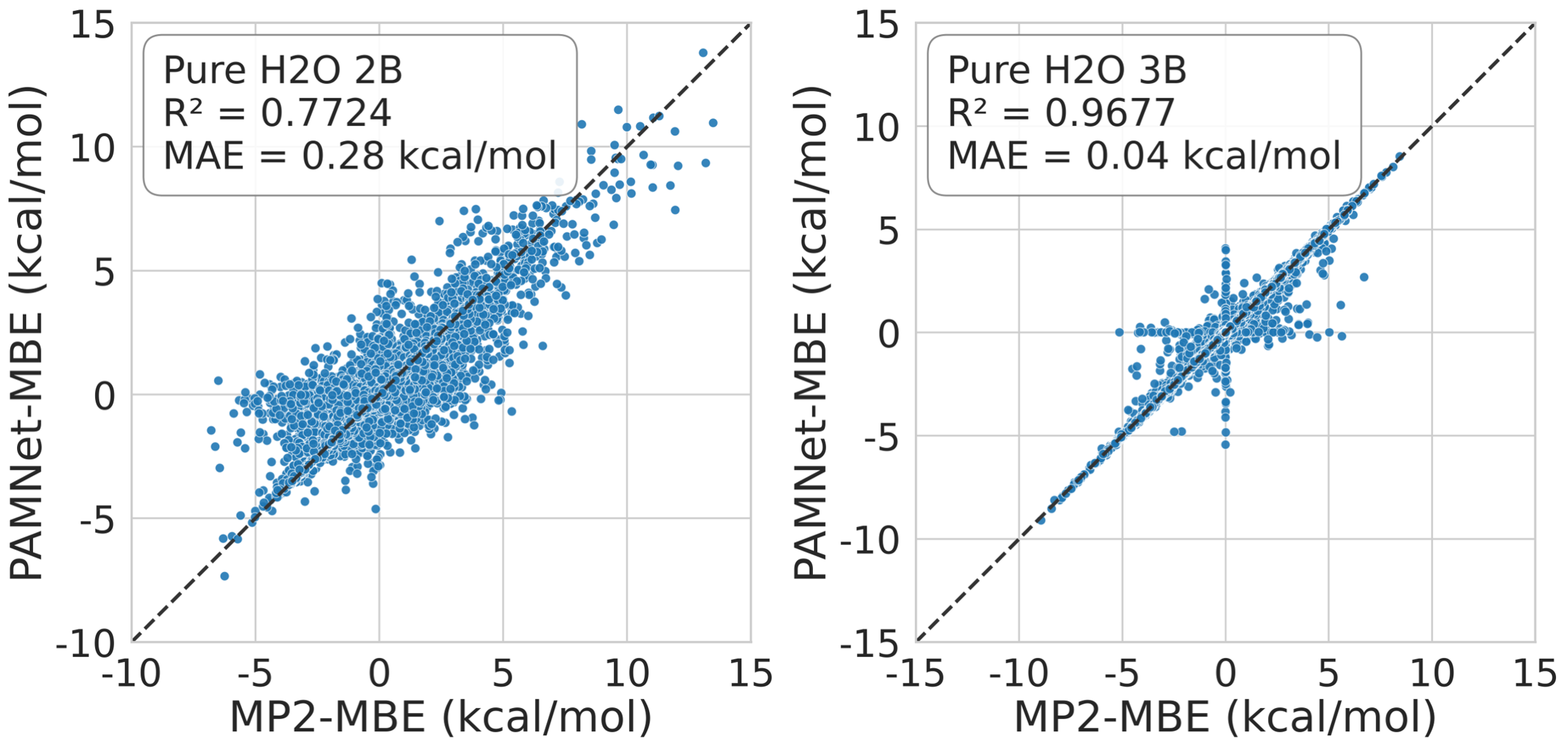
Figure 7: 2B and 3B energies in mixed-density water clusters, demonstrating improved generalization of PAMNet-MBE via multi-stage training.
Generalization to PES and Dissociation Curves
Fine-tuned PAMNet-MBE accurately reproduces 1D dissociation curves for water and phenol dimers, closely matching MP2 or DFT references in both shape and well depth—demonstrating robust encoding of both quantum hydrogen bonding and π–π stacking interactions.
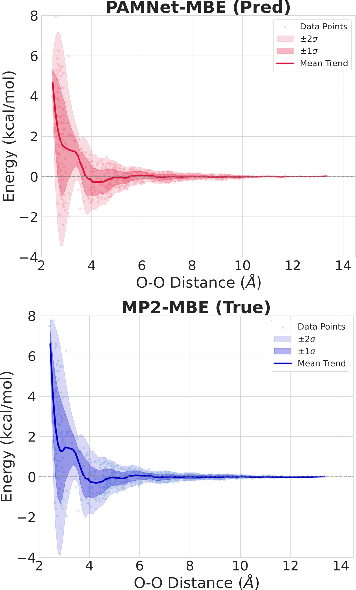
Figure 8: 1D dissociation curves of all water dimers in double-density N0 as a function of O–O distance: PAMNet-MBE (top) vs. MP2-MBE (bottom).
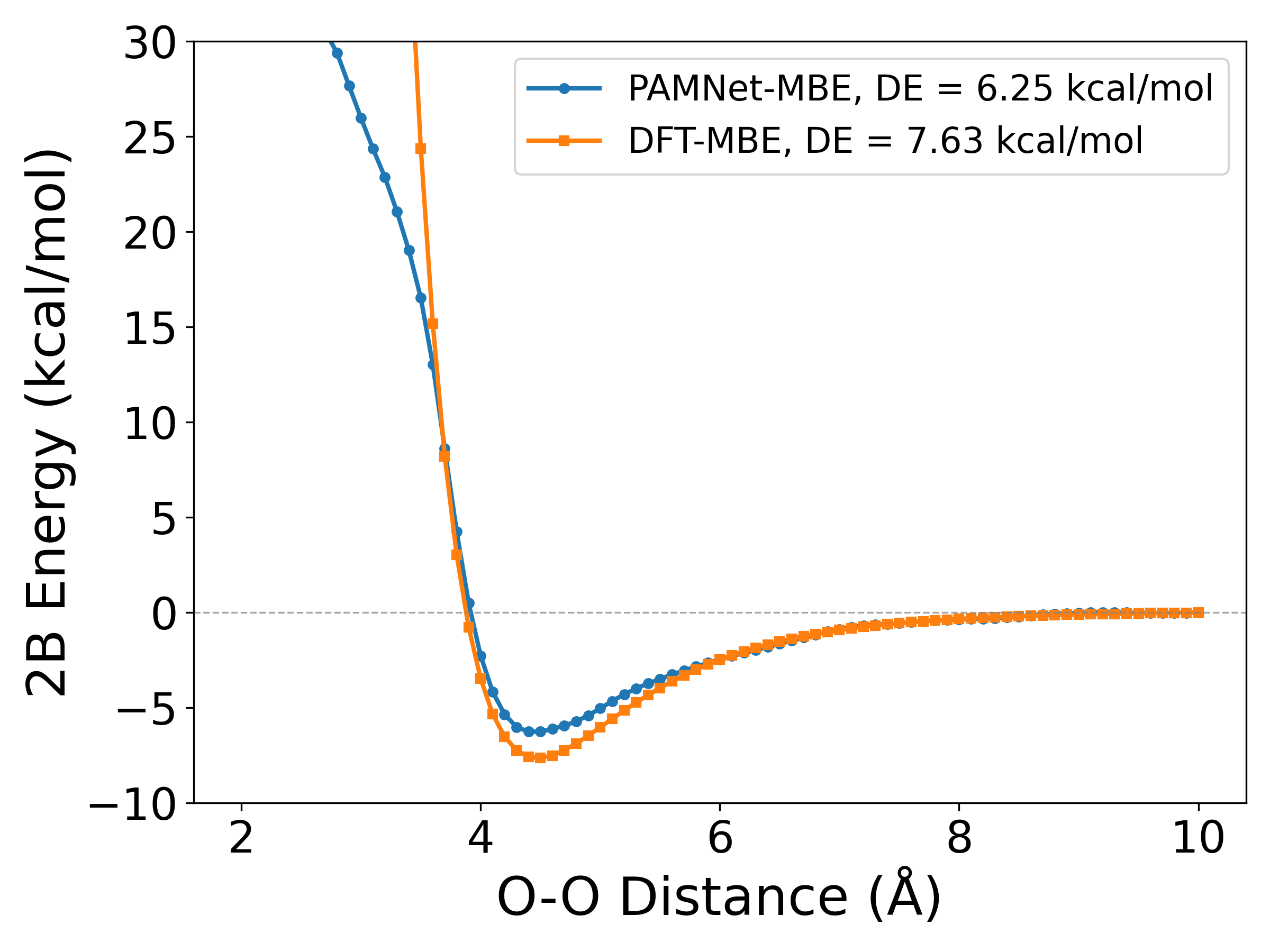
Figure 9: 1D PES of a phenol dimer (O–O axis): PAMNet-MBE versus DFT-MBE. Dissociation energies in close agreement.
Data-Efficient Transfer via Knowledge Distillation
The teacher–student distillation protocol enables light-weight GNNs to retain high accuracy on unseen cluster sizes after minimal fine-tuning, even outperforming direct model transfer or naive fine-tuning. Models such as DimeNet and ViSNet (with explicit angular and vector basis) generalize best to small water clusters, recovering both energy and physical trends, and converging quickly with only a handful of target samples.
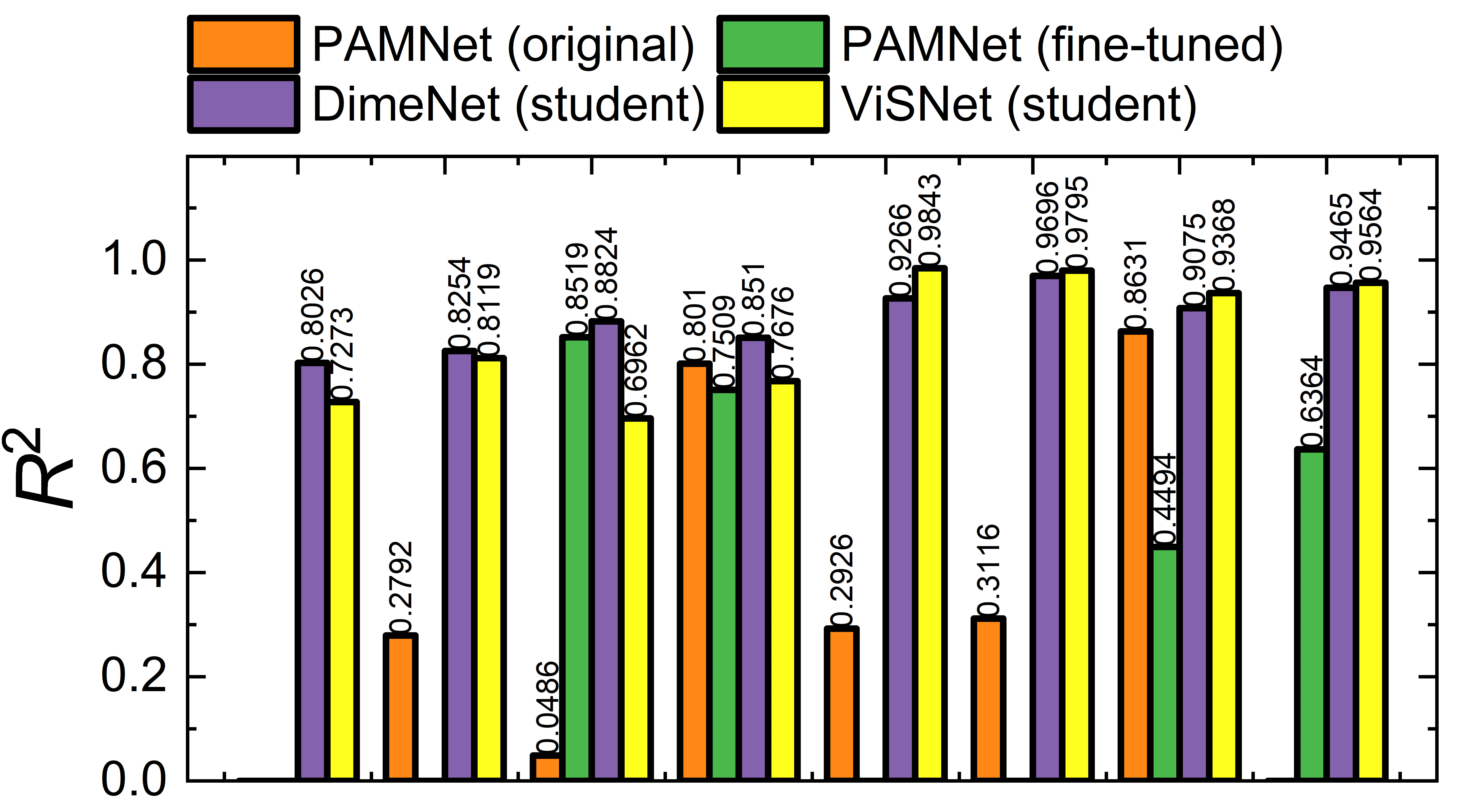
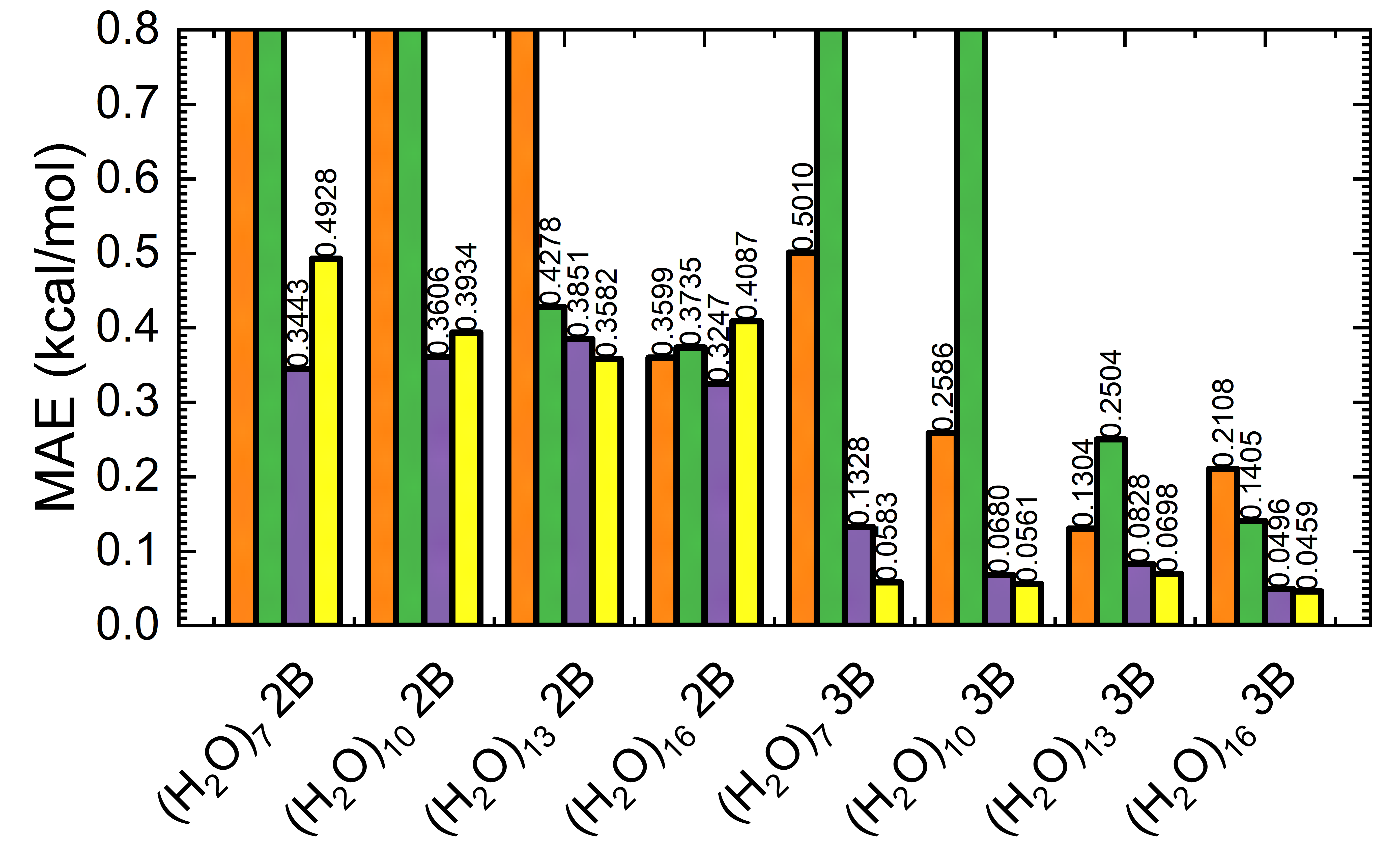
Figure 10: 2B (top) and 3B (bottom) energy metrics on normal-density small water clusters: comparison of original, fine-tuned teacher, and student (DimeNet/ViSNet) models.
Implications and Future Directions
Practical Implications
- Large-Scale Molecular Simulation: FB-GNN-MBE enables fast and accurate PES prediction for systems previously intractable to ab initio simulation, facilitating quantum-fidelity molecular dynamics and structure exploration in large aqueous/heterogeneous systems.
- Model Transferability: The data-efficient transfer afforded by teacher–student distillation protocol is particularly impactful for extending pretrained potentials to new chemistries, cluster sizes, or environmental conditions with minimal retraining.
Theoretical Implications and Perspectives
FB-GNN-MBE highlights the importance of chemical hierarchy and fragment distinction in molecular ML, providing a physically grounded alternative to traditional NN or GNN PES surrogates. The hierarchical and physically informed message-passing networks implemented here can be generalized further, potentially to reactive systems (with dynamic bonding), polarizable force fields, or electronically excited states.
Future methodological extensions may include:
- Application to systems with covalent bond rearrangement (requiring bond-cleaving fragmentation and adaptive graphs),
- Multi-task architectures for joint energy/force learning and energy decomposition analysis,
- Integration with meta-learning for rapid adaptation to completely unseen chemistry.
Conclusion
FB-GNN-MBE offers a scalable, accurate, and interpretable framework for PES prediction in chemically heterogeneous and hierarchically structured systems. By marrying QM-based MBE with fragment-based GNNs and leveraging knowledge distillation for transferability, the methodology outperforms conventional ML and GNN potentials across a variety of chemical environments. Its combination of chemical accuracy, computational efficiency, and transfer learning capability positions it as a powerful tool for quantum-mechanical modeling of large molecular assemblies and paves the way for further advances in data-driven many-body electronic structure theory.