- The paper constructs a richly annotated imbalanced dataset from ENCODE to benchmark ML-driven quality control in diverse NGS assays.
- It employs two orthogonal feature sets (QC-34 and BL) and demonstrates high classifier performance, with RNA-Seq AUC ROC exceeding 0.9.
- The study facilitates research in feature selection, dimensionality reduction, and bias analysis, providing open-source tools for immediate application.
An Imbalanced ENCODE-Based Dataset with Multi-Granular Feature Representations for Quality Control in NGS
Introduction
The proliferation of next-generation sequencing (NGS) techniques has catalyzed advances in genomics, but the downstream utility of these data critically depends on rigorous quality control (QC). The complexity and volume of NGS experiments introduce heterogeneous sources of noise and systematic error, rendering manual QC infeasible at scale. Existing genomic consortia such as ENCODE provide quality labels and some metrics, but the curation, normalization, and tabularization of features suitable for robust machine learning remains insufficient, impeding the development and benchmarking of automated QC solutions.
The paper "An Imbalanced Dataset with Multiple Feature Representations for Studying Quality Control of Next-Generation Sequencing" (2604.04981) systematically addresses this gap by constructing and validating a large-scale, richly annotated tabular dataset derived from 37,491 human and mouse NGS samples in ENCODE. This resource is explicitly structured for ML-driven QC, offering two orthogonal feature representations and detailed metadata to facilitate multi-dimensional benchmarking.
Dataset Generation and Feature Construction
The authors curated all released and revoked human and mouse NGS samples (covering ChIP-Seq, RNA-Seq, DNase-Seq, and eCLIP assays) from ENCODE, filtering for unequivocal QC labeling and excluding ambiguous or archived data. The final dataset maintains the original experimental and biological diversity, with an overall 3.2% prevalence of revoked (low-quality) samples.
The feature generation pipeline is depicted as follows:
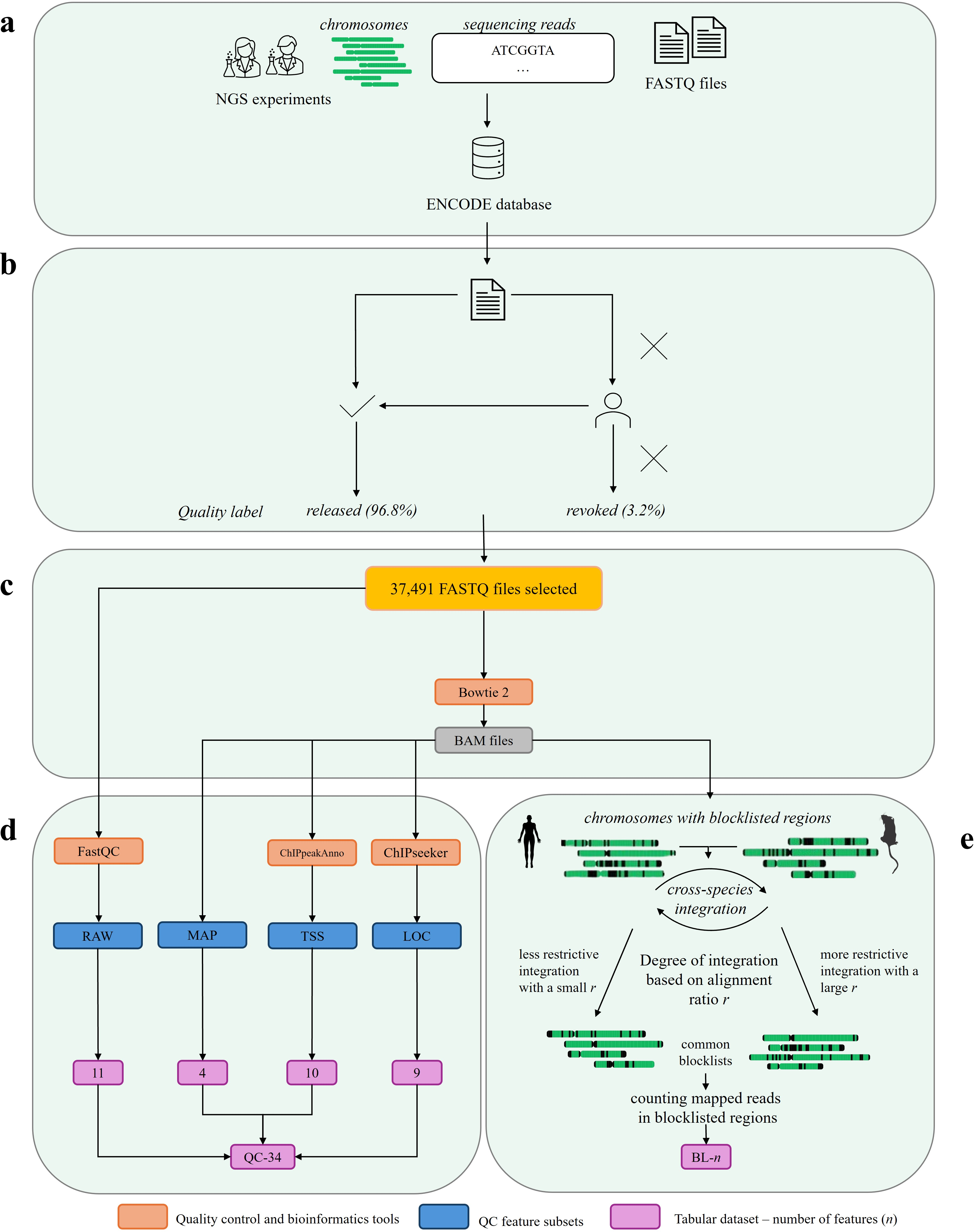
Figure 1: Overview of the data and feature generation workflow, illustrating data acquisition, QC labeling, alignment, and computation of both QC-34 and BL features.
Two main classes of features are available:
- QC-34: Thirty-four interpretable metrics synthesized from established QC and bioinformatics tools, partitioned into RAW (ordinal outcomes from FastQC flags), MAP (Bowtie 2-based mapping statistics), TSS (read density around transcription start sites), and LOC (distribution across genome functional elements). The distributions and relationships of these features are elaborated as:
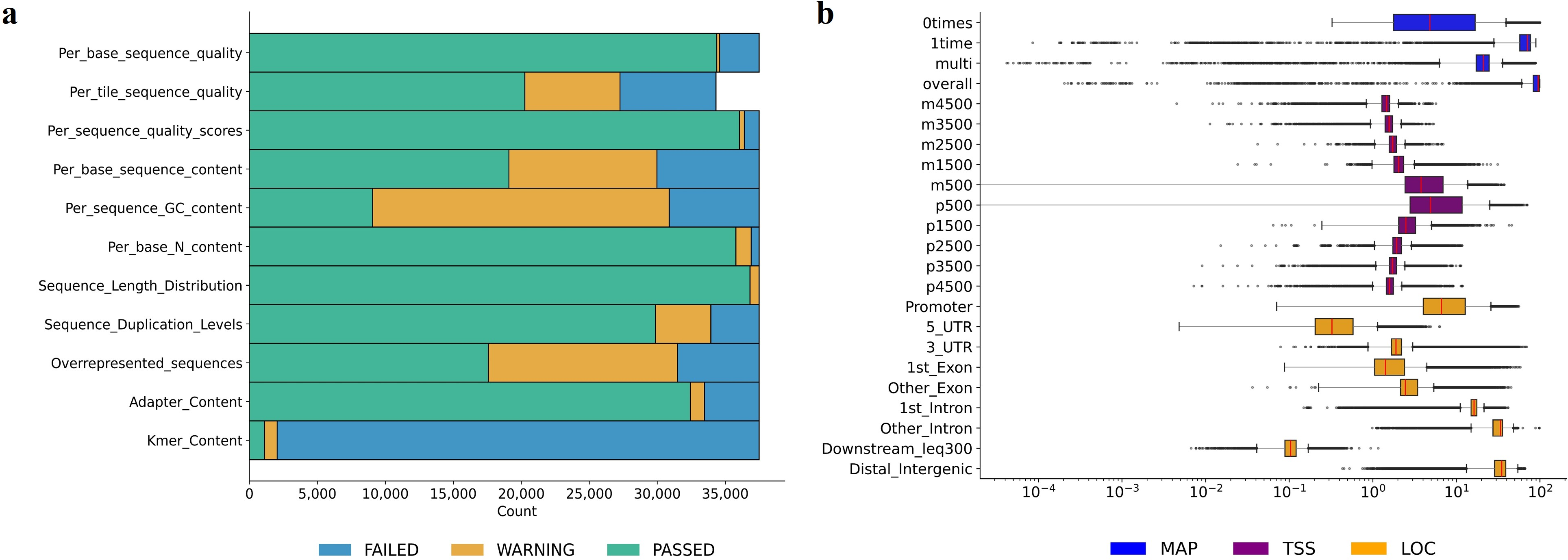
Figure 2: Empirical distributions of RAW features (ordinal) and MAP, TSS, and LOC features (numeric, log-scale) provide insights into data heterogeneity and potential for discriminative modeling.
- BL (Blocklist): Features enumerating the number of reads in merged blocklisted regions identified via ENCODE blacklist annotations. The authors devised a unified human-mouse blocklist via cross-species mapping (liftOver) and implemented an alignment ratio threshold for inclusion, controlling feature dimensionality between 8 and 1,183 and enabling analyses spanning varying biological orthology and curse-of-dimensionality regimes.
The growth trajectory in BL feature set cardinality as the alignment ratio is relaxed is visualized below:
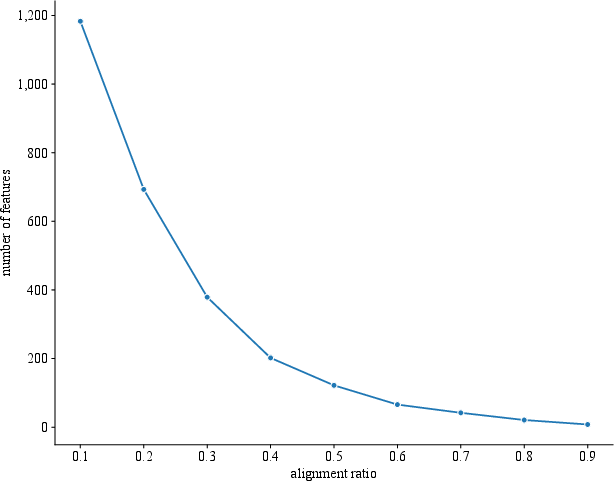
Figure 3: Relationship between alignment ratio stringency and the number of BL features; low stringency admits additional, species-specific problematic regions.
Technical Validation and Supervised Benchmarks
Extensive validation is performed on both the label and feature quality:
Among notable findings:
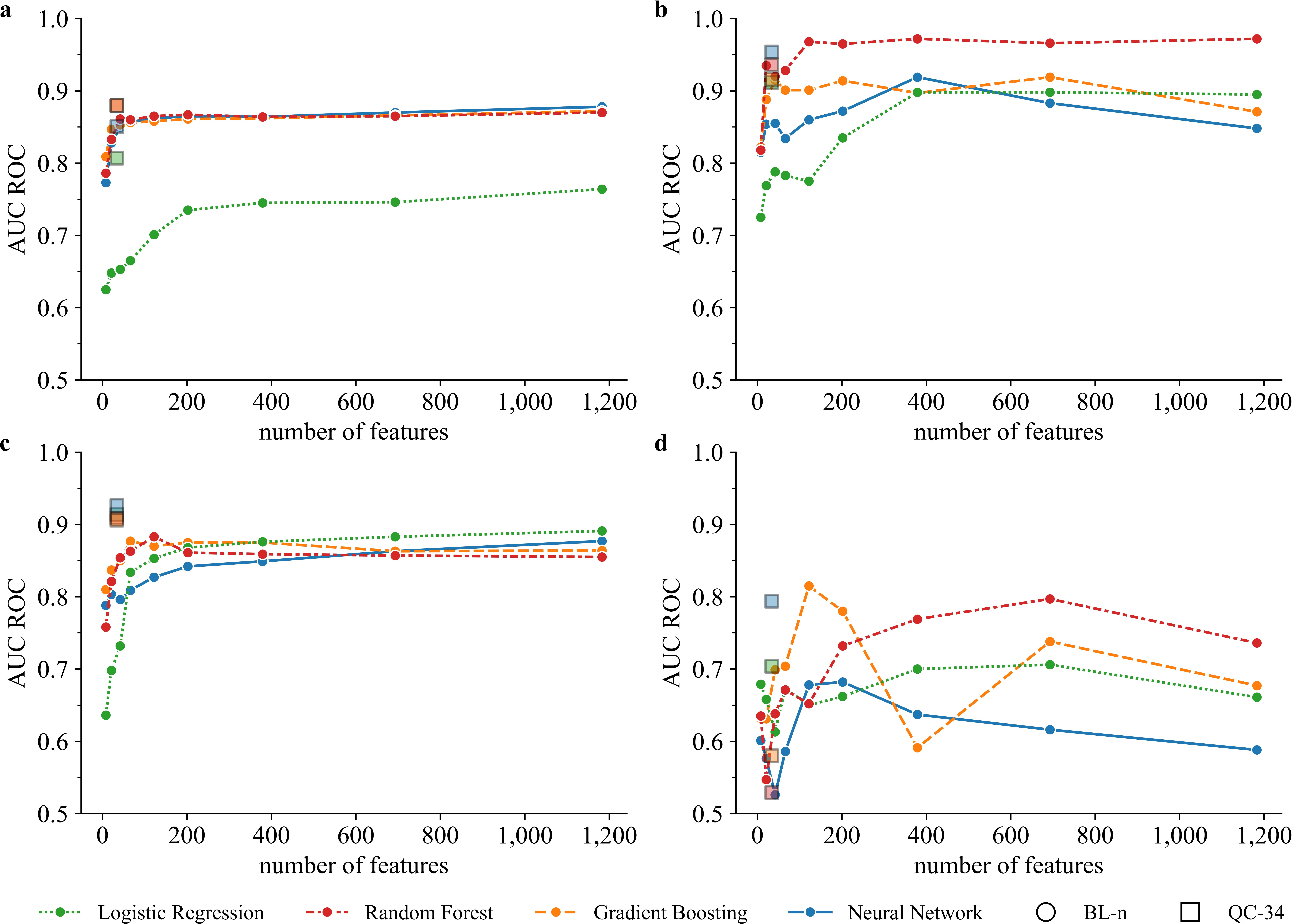
- For ChIP-Seq and DNase-Seq, RF, GB, and NN achieve AUC ROC > 0.7 across almost all BL feature subsets and QC-34.
- In RNA-Seq, AUC ROC exceeds 0.9 for QC-34 and also for selected BL feature regimes, emphasizing the strong discriminatory power for transcriptomic QC.
- Classifier performance generally saturates when the BL feature set grows beyond ~200 features, empirically demonstrating the practical onset of the curse of dimensionality and redundancy—especially relevant for high-dimensional anomaly detection tasks.
- For eCLIP assays, all approaches exhibit lower and more unstable performance, signifying assay-specific QC challenges and suggesting the need for further feature engineering or context-aware modeling.
Spectrum of Use Cases and Implications
The dataset's multi-representational design makes it highly amenable to several research directions:
- Benchmarking Feature Selection and Dimensionality Reduction: Because BL feature sets are nested, this resource supports systematic evaluation of biological and statistical feature selection algorithms, including hybrid and multi-view learning strategies, within a controlled biological context.
- Algorithm Robustness and Generalization: Stratified assays and detailed metadata facilitate the evaluation of cross-assay generalization, batch effects, and robustness to label noise, critical in biomedical ML applications.
- Curse of Dimensionality Studies: Variable BL granularity allows controlled investigations into model performance trade-offs, regularization, and outlier/anomaly detection strategies in high-dimensional bioinformatics, as shown by the performance plateauing beyond 200 features.
- Bias and Demographic Analysis: Associated metadata on donor demographics and experiment structure underpin fairness and bias analyses, though users are cautioned regarding representativeness limitations, especially regarding ethnic diversity.
The ready availability of Python and R code, as well as tabular CSVs and metadata on Zenodo, further facilitates direct adoption for benchmarking, model development, or integration into larger multi-omic studies.
Limitations and Directions for AI-Driven Genomics
The authors note several caveats. Demographic imbalances inherent to the original ENCODE depositions may bias downstream AI models. QC labels—though externally validated and augmented by expert curation—remain imperfect, especially for "released" class samples. Certain NGS modalities (e.g. single-cell RNA-Seq) are not yet covered, and periodic dataset updates are planned to address shifts in labeling or assay representation.
From a future AI perspective, this dataset provides not only a gold standard for developing ensemble and deep learning-based QC pipelines, but also a reference for transfer learning, active learning, and outlier detection research in genomics. The nested, orthogonal feature construction enables modularity in representation learning and the testing of semi-supervised and unsupervised anomaly detection under realistic class imbalance scenarios.
Conclusion
This work establishes a robust, scalable, and deeply annotated resource for machine learning-driven quality control of NGS data, bridging multiple representation paradigms and providing rigorous external validation and technical benchmarking. The focus on both technically agnostic (QC-34) and genome-informed (BL) features spanning variable dimensionalities enables a wide spectrum of algorithmic inquiries, from feature selection to robustness and fairness evaluation. The integration with public repositories and codebases ensures sustained impact and adaptability for the computational genomics and AI communities.