A New Paradigm for Computational Chemistry
Abstract: Computational chemistry has become an indispensable tool for generating data and insights, pervading all branches of experimental chemistry. Its most central concept is the potential energy hypersurface, key to all chemistry and materials science, as it assigns an energy to a molecular structure, the necessary ingredient for reaction mechanism elucidation and reaction rate calculation. Density functional theory (DFT) has been the most important method in practice for obtaining such energies, which is mirrored in the use of high-performance computing hardware. In the last two decades, a new class of surrogate potential energy functions has been evolving with remarkable properties: quantum accuracy combined with force-field speed. Until very recently, their application was hampered by the fact that they needed to be trained on truly large system-specific data sets, generated before a computational chemistry study could be started (in sharp contrast to DFT, which, as a first-principles method, works out of the box, but at a far higher price of computational cost). Very recently, this roadblock has been overcome by so-called foundation machine learning interatomic potentials, which are poised to completely change the way we do computational chemistry, likely prompting us to abandon DFT as the prime method of choice for this purpose in less than a decade.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper explains a big shift happening in computational chemistry. For decades, chemists have used a method called density functional theory (DFT) to calculate the “energy landscape” of molecules—think of a 3D map of hills and valleys that tells you which structures are stable and how hard it is to move from one to another. DFT is accurate but slow and expensive.
The authors describe a new family of machine-learning models—machine learning interatomic potentials (MLIPs)—that can predict these energies much faster, while staying close to quantum‑level accuracy. Even more exciting, a new generation called “foundation MLIPs” can often work “out of the box” without retraining for each new system. The paper argues these models are likely to become the default tool for many chemistry problems within a decade, possibly replacing DFT as the first choice.
What questions are the authors trying to answer?
They focus on a few simple questions:
- Can ML models predict molecular energies and forces as accurately as quantum methods, but much faster?
- Can we build general “foundation” models that work on many different molecules and materials without retraining?
- When should we use these models as-is, and when should we fine‑tune them for a specific problem?
- How fast are these models in practice, and how do they compare with traditional methods?
- What are the current weaknesses, and what needs to be improved so we can trust them in real research?
How did they approach the problem?
This is a review and perspective paper, not a single experiment. The authors:
- Explain how modern ML potentials are built and why they respect basic physics (like not caring if you rotate or move a molecule).
- Summarize the evolution from early, system‑specific models to today’s large “foundation MLIPs” trained on huge, varied datasets.
- Compare speeds in a small timing test, and discuss trade‑offs between using a general model “as is” versus fine‑tuning it for a specific task.
- List the main challenges (like handling long‑range forces, magnetism, and trustworthy uncertainty estimates) and the most promising solutions.
From DFT to MLIPs: the basic idea
- Think of the energy landscape like a terrain map. DFT measures the height (energy) at any point very precisely—but it’s slow, like surveying each spot by hand.
- MLIPs learn the shape of the terrain from many examples, so they can “guess” new heights very quickly—like a video game engine that renders realistic physics in real time.
How MLIPs “see” molecules
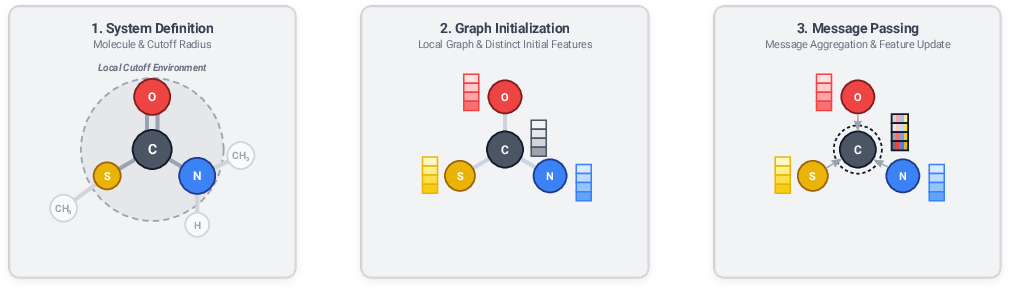
- Symmetry matters: If you rotate, shift, or swap identical atoms, the true energy shouldn’t change. MLIPs are designed to respect that.
- Local neighborhoods: Many MLIPs split the total energy into atomic contributions based on nearby atoms (like each atom listening to its neighbors within a certain distance). This keeps models fast and able to handle big systems.
- Graphs and message passing: A molecule is turned into a graph (atoms = dots, bonds/nearby pairs = lines). Atoms “message” their neighbors and update their internal features over several rounds, learning how local structure affects energy and forces.
- Equivariance: Some models keep track of directions (not just distances). That helps predict not only energies (which don’t depend on direction) but also vector/tensor properties like forces or dipoles in the correct orientation.
Foundation models for chemistry
- Like big LLMs trained on lots of text, foundation MLIPs are trained on massive collections of molecules and materials.
- After this pretraining, many can be used directly on new systems. Some well-known examples include MEGNet, M3GNet, MACE‑MP‑0, eqV2, and UMA.
- These models vary in size and training data. The trend is clear: bigger, broader datasets → better general skills and fewer retraining needs.
A small speed check
The authors optimized the structures of a simple molecule (naphthalene) using different methods and found:
- Classical force fields (older, hand‑crafted physics): fastest.
- MLIPs: much slower than force fields, but hundreds to a thousand times faster than DFT in this test.
- DFT: slowest by far.
Takeaway: MLIPs sit in the sweet spot—much faster than DFT and much more accurate than basic force fields.
What did they find, and why does it matter?
Here are the main takeaways.
- Foundation MLIPs work surprisingly well out of the box
- Many modern models can predict energies and forces for a wide range of materials and molecules without retraining.
- They can handle tasks like running long molecular dynamics simulations and screening materials at scale.
- Speed and accuracy are game‑changing
- MLIPs can be orders of magnitude faster than DFT while often reaching DFT‑level accuracy.
- This makes it possible to study larger systems, explore more possibilities, and do it all in a reasonable time.
- Generalist vs. fine‑tuned use
- Using a model “as is” is easy and avoids extra training time.
- Fine‑tuning can improve accuracy for a specific system but risks “forgetting” older knowledge (called catastrophic forgetting).
- New techniques (like LoRA adapters and knowledge distillation) help adapt or shrink models while keeping what they’ve learned.
- Reliable “I’m not sure” signals are needed
- Many workflows need an uncertainty estimate—basically, error bars.
- Ensembles are great but not always available for one big released model.
- Practical, add‑on uncertainty methods exist, but more built‑in, well‑calibrated solutions are needed.
- Current limits and active fixes
- Long‑range interactions (like electrostatics and dispersion) are hard if a model only “listens” locally; new designs combine local learning with long‑range physics or attention mechanisms.
- Charge and magnetism: total charge and spin are being added, but detailed magnetic behavior (like antiferromagnetism) is still challenging in general‑purpose models.
- Advanced quantities (like Hessians for transition states) are expensive to compute even for ML models; methods are being developed to make this practical.
- Data quality matters: very large datasets can contain noisy or inconsistent entries, so better curation and “minimal, high‑value” datasets are important.
Why it matters: If MLIPs can be trusted across many domains, chemists will be able to model and explore chemistry at scales and speeds that were out of reach with DFT. That means faster discovery of materials, catalysts, drugs, and more.
What could this change in the future?
- DFT may stop being the default starting point
- For many problems, people may begin with a foundation MLIP, only calling DFT or higher‑level quantum methods to check or improve uncertain cases.
- Over time, training MLIPs on very accurate quantum data (beyond DFT) could make them even better than today’s DFT in both speed and accuracy.
- Bigger, more realistic simulations become routine
- MLIPs make it practical to simulate large, complex systems (like porous materials, polymers, and biomolecules) with near‑quantum accuracy.
- This bridges the gap between “quantum chemistry” and “materials/biomolecular modeling.”
- Better workflows and education
- Uncertainty estimates and automatic fine‑tuning will be built into standard tools, making advanced simulations easier and safer to use.
- Chemistry training will shift to include more data‑driven and AI‑based modeling.
In short, the paper argues that machine‑learning potentials are on track to transform computational chemistry—from how we compute energies to how we design materials—by delivering quantum‑grade insights at a fraction of the time and cost.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
The following list distills the concrete gaps and unresolved questions highlighted or implied by the paper that future work should address:
- Long‑range interactions: No consensus on the best strategy (physics‑driven vs learned) to capture electrostatics, polarization, and dispersion beyond a local cutoff; need systematic, apples‑to‑apples evaluations of MACE‑POLAR‑style explicit physics, LES/Qeq‑type latent charge schemes, nonlocal descriptors, and all‑to‑all attention (e.g., AllScAIP), including accuracy/complexity trade‑offs, scalability under PBC, and robustness to metallic screening.
- Scaling with long‑range terms: Efficient algorithms (e.g., FMM, particle‑mesh Ewald, sparse attention) to preserve near‑linear scaling when adding nonlocal interactions are not established for foundation MLIPs.
- Charge and spin conditioning: Many foundation MLIPs lack explicit conditioning on total charge and spin multiplicity; even fewer support local magnetic moments (collinear/non‑collinear) or spin textures. Methods to input, predict, and generalize across unseen spin configurations and charge states remain to be developed and validated.
- Relativistic and heavy‑element coverage: Systematic strategies (data, architectures, and labels) for f‑block/heavy‑element chemistry, spin‑orbit coupling, and magnetocrystalline anisotropy are missing.
- Excited states and non‑adiabatic dynamics: Foundation MLIPs largely target ground‑state PESs; scalable approaches to excited‑state PESs, non‑adiabatic couplings, and conical intersections (and their uncertainty) remain open.
- Surfaces and interfaces: Bulk‑trained models underperform on surfaces/adsorption; curated, high‑quality surface and interface datasets (with consistent DFT settings and coverage of steps, kinks, and reconstructions) and benchmarks are needed to close this gap.
- Reactive chemistry coverage: Reliable TS search, reaction path sampling, and barrier prediction with foundation MLIPs require (i) training with information beyond energies/forces (e.g., Hessians), (ii) robust validation on reaction benchmarks (gas phase and condensed phase), and (iii) protocols for detecting mechanistic failure modes.
- Hessians at scale: Memory‑ and time‑efficient training and inference for second (and higher) derivatives with large equivariant architectures are unresolved; practical adjoint/forward‑mode strategies, mixed precision, and block‑sparse implementations are needed.
- Vibrational properties: Systematic evaluation of how Hessian‑augmented training improves phonons, IR/Raman, and anharmonicity (including third/fourth derivatives) is missing for foundation MLIPs.
- Stress/virial consistency: Standardized support for stress tensors/virials (and their derivative consistency with energy/forces) for NPT simulations is not yet established across models.
- Conservative forces and MD stability: Some models predict forces directly; rigorous guarantees of energy conservation, long‑time MD stability, and drift bounds (with and without thermostats/barostats) are lacking.
- Application‑level benchmarks: Community‑accepted, task‑driven benchmarks (MD stability over long horizons, TS‑location success rates, diffusion constants, adsorption energies, surface energies, phase transitions) with fixed protocols and reference levels are still limited (MLIP Arena is a start but incomplete).
- Speed/accuracy/energy metrics: No standardized, hardware‑aware benchmark suite that reports accuracy, wall‑time, and power consumption versus system size across CPUs/GPUs/accelerators; reproducible harnesses are needed to compare MLIPs, FFs, and DFT fairly.
- Continual learning without forgetting: Methods to fine‑tune foundation MLIPs while preventing catastrophic forgetting in equivariant architectures (e.g., equivariant LoRA, replay, parameter isolation) need systematic, large‑scale evaluation and best‑practice protocols.
- Knowledge distillation pipelines: General recipes to distill foundation MLIPs into faster, system‑specific students (with provable or empirical guarantees on accuracy and stability in target domains) are not yet standardized.
- Uncertainty quantification (UQ): Most foundation MLIPs ship without integrated, calibrated UQ; scalable single‑model UQ (beyond ensembles), calibration under distribution shift, and task‑aware UQ (e.g., for MD, TS, surfaces) remain open.
- Post‑hoc UQ at scale: Distance‑based UQ requires training data access; gradient‑based UQ requires a one‑pass over massive datasets. Efficient, privacy‑preserving, and compute‑light post‑hoc UQ for billion‑example models is unresolved.
- OOD detection and guardrails: Reliable, thresholded OOD detectors (that trigger fine‑tuning or ab initio fallback) with low false‑negative rates in real workflows are missing.
- Dataset quality control: Automated, scalable QC to detect noisy DFT labels (e.g., non‑zero net forces), inconsistent settings, duplicates, and unphysical structures is not yet standard for multi‑million to billion‑entry corpora.
- Data efficiency and coverage: Principles for constructing minimal, maximally informative datasets (active learning across domains, diversity/novelty metrics, coverage of high‑energy/short‑range repulsion) are underdeveloped for foundation training.
- Multi‑fidelity training: Robust frameworks to mix DFT flavors and high‑level ab initio (e.g., Δ‑learning to CC/NEVPT2) without “functional confusion,” with clear target levels and calibration to experiment, are needed.
- Path to ab initio labels at scale: Practical strategies to inject limited coupled‑cluster/multireference labels (where they matter most) into foundation training, including selection policies and transfer‑learning schedules, are not established.
- Short‑range physics and robustness: Guarantees against unphysical behavior at close approaches (e.g., missing Pauli repulsion) under extrapolation are not formalized; physics‑informed constraints or shielding strategies need evaluation.
- Periodic boundary conditions and charged cells: Unified treatments for charged systems under PBC, neutralizing backgrounds, long‑range corrections, and metallic screening within MLIPs remain incomplete.
- Thermodynamics and free energies: Protocols to propagate MLIP/UQ uncertainty to free energy estimates (alchemical/metadynamics) and to assess bias versus DFT/experiment are lacking.
- Multiscale coupling: General, stable schemes for QM/MLIP/MM adaptive embedding (charge consistency, polarization, and error control at boundaries) are not yet mature.
- Property multitask learning: Unified training to jointly predict energies, forces, charges, dipoles/polarizabilities, stress, and other tensorial observables with consistent equivariance and manageable cost needs principled design and ablation studies.
- Efficient high‑order equivariance: Architectures supporting higher‑rank tensors without prohibitive Clebsch–Gordan costs (or robust Cartesian alternatives) remain an open design challenge.
- Interpretability and chemical concepts: Methods to extract reliable chemical insight (attributions, learned descriptors, reaction coordinates) from foundation MLIPs are nascent and unvalidated across domains.
- Element/chemistry coverage maps: Clear, published capability maps (which elements, charge/spin states, bonding motifs, and thermodynamic ranges are in‑ or out‑of‑scope for each model) are generally missing.
- Software provenance and reproducibility: Standardized reporting of model versioning, training data lineage, and uncertainty‑aware defaults is not yet common, risking silent regressions in practice.
- Environmental cost: Transparent accounting and reduction strategies for the carbon footprint of training/inference of billion‑parameter MLIPs, relative to traditional DFT workloads, are not provided.
Practical Applications
Immediate Applications
The paper’s findings and methods enable a set of deployable use cases across sectors today, largely by substituting foundation MLIPs for routine DFT in many computational workflows and by introducing practical strategies for fine-tuning, uncertainty quantification, and acceleration.
Industry (Chemicals, Materials, Energy, Semiconductor)
- High-throughput prescreening of materials at near-DFT fidelity
- Use case: Rank candidate inorganic solids (e.g., ~10k+ semiconductors) for lattice dynamics, stability, and phonon properties before targeted DFT/experiment.
- Tools/workflows: MatterSim v1, MACE-MP-0; pipeline via ASE/LAMMPS + GPU nodes; job arrays on cloud/HPC.
- Dependencies/assumptions: Target chemistry represented in training sets (e.g., Materials Project-derived data); validate critical candidates with DFT; GPU availability; error thresholds acceptable for go/no-go decisions.
- Rapid molecular dynamics (MD) for materials process design
- Use case: Stable MLIP-driven MD to evaluate phase behavior (e.g., silica polymorph transitions; zeolite metastability) and liquid behavior (e.g., water) without system-specific retraining.
- Tools/workflows: Foundation MLIPs deployed in MD engines; optional knowledge distillation to smaller students for faster MD in narrow chemical spaces.
- Dependencies/assumptions: Validation on target temperature/pressure ranges; foundation model stability in OOD regimes; MD failure cases still occur even with low force error—benchmark on MLIP Arena-like tasks.
- Catalyst and surface chemistry prototyping with targeted fine-tuning
- Use case: Estimate adsorption energetics and reaction intermediates on metal surfaces for catalyst leads; fine-tune foundation MLIPs where surface energies/adsorption are inadequately captured.
- Tools/workflows: UMA/eqV2 baseline + LoRA or lightweight head-adaptation; active learning loop selecting small DFT/ab initio batches; integrated UQ to drive data acquisition.
- Dependencies/assumptions: Risk of catastrophic forgetting—prefer LoRA/readout heads over full fine-tunes; surface and adsorbate coverage in pretraining may be limited; maintain replay/regularization where broader generality must be kept.
- Battery and energy materials screening (electrolytes, MOFs, solid electrolytes)
- Use case: Screen transport pathways, lattice stability, and host–guest interactions in porous/ionic solids at scale before experiment.
- Tools/workflows: MLIPs with long-range augmentation when needed (e.g., MACE-POLAR-1, AllScAIP); Ewald/LES/Qeq-based add-ons; automated workflow managers.
- Dependencies/assumptions: Long-range electrostatics/dispersion critical; if the chosen MLIP lacks long-range physics, augment or switch; computational scaling can increase to O(N log N)–O(N²).
- Carbon capture and separations material ranking
- Use case: Prioritize MOFs/molecular crystals for adsorption selectivity and stability using approximate PES at MLIP speed.
- Tools/workflows: UMA/all-to-all attention MLIPs; GPU-accelerated high-throughput pipelines; UQ thresholds for escalation to DFT/experiment.
- Dependencies/assumptions: Foundation model coverage of relevant chemistries; adsorption in pores may necessitate long-range-aware models and small targeted fine-tunes.
Pharma/Healthcare
- Fast conformer ranking for organometallic and small-molecule leads
- Use case: Rank conformers of transition-metal complexes and drug-like molecules to guide docking and QM follow-up.
- Tools/workflows: UMA-S-1p2 or similar foundation MLIP in conformer generators; fallback to DFT when close energy spacings (< few kJ/mol).
- Dependencies/assumptions: Reliability drops for highly flexible/fluxional systems and near-degenerate conformers; verify with small DFT set.
- Accelerated property prediction within physics-aware pipelines
- Use case: Approximate dipoles, vibrational frequencies, and derived properties through MLIP energies/forces for screening and QSAR features.
- Tools/workflows: Equivariant MLIPs; finite-difference frequencies; force-as-gradient architectures to ensure tensor consistency.
- Dependencies/assumptions: Hessians are costly; for vibrational tasks, validate on small calibration sets; consider models trained with Hessian signals when available.
Software & HPC
- Replace routine DFT steps with MLIPs to reduce compute cost and emissions
- Use case: Substitute DFT energy/force evaluations in optimization/MD loops; reduce job durations by orders of magnitude.
- Tools/workflows: ASE + foundation MLIPs on GPUs; use geometric tolerances consistent with MLIP noise; track energy budgets.
- Dependencies/assumptions: Accuracy tolerances matched to use; CPU-only environments may see less gain; careful software integration and model compatibility.
- Knowledge distillation for targeted production workloads
- Use case: Train compact student MLIPs for a narrow chemical subspace to achieve force-field-like speed with near-teacher accuracy.
- Tools/workflows: Teacher–student distillation pipelines; deploy students in MD/high-throughput loops; formal performance SLAs.
- Dependencies/assumptions: Domain restriction clearly defined; periodic revalidation against teacher/DFT; distillation can miss rare-event physics.
- Post-hoc uncertainty quantification for single-model deployments
- Use case: Flag out-of-scope predictions in production without retraining.
- Tools/workflows: Gradient-based sensitivity measures; feature-space distance methods with small reference caches; readout ensembling where heads can be added.
- Dependencies/assumptions: Calibration with a small, task-relevant DFT set is recommended; distance-based methods require access to training embeddings or curated reference sets.
Academia & Education
- Course and lab modernization
- Use case: Teach PES and molecular simulation labs using foundation MLIPs to enable realistic systems on modest GPUs.
- Tools/workflows: Jupyter/Colab + ASE; model zoo with curated examples (molecules, crystals, surfaces).
- Dependencies/assumptions: Open models and permissive licenses; clear guidance on uncertainty and when to escalate to DFT.
- Benchmarking and method development at scale
- Use case: Evaluate robustness of MLIPs on application-like tasks (e.g., MD stability, TS searches, phonons) beyond MSE metrics.
- Tools/workflows: MLIP Arena benchmarks; standardized task suites for MD/phonons/transition states; open leaderboards.
- Dependencies/assumptions: Shared datasets with clean metadata and QA; community agreement on acceptance thresholds.
Policy & Governance
- Green computing initiatives in computational chemistry
- Use case: Adopt MLIP-first policies for exploratory screening to reduce HPC energy consumption, reserving DFT/CCSD(T) for confirmatory steps.
- Tools/workflows: Procurement guidelines prioritizing GPU-efficient MLIP workflows; monitoring dashboards of CO₂e savings.
- Dependencies/assumptions: Availability of validated MLIP pipelines; governance on uncertainty thresholds and escalation criteria.
- Data quality and curation standards
- Use case: Enforce automated QA for training data (e.g., net-force checks, DFT parameter sanity) in public/consortia datasets.
- Tools/workflows: CI pipelines that reject noisy calculations; dataset “model cards” documenting functional choices and known caveats.
- Dependencies/assumptions: Community buy-in; resourcing for data stewardship; reproducibility infrastructure.
Daily Life and Product R&D
- Faster materials-enabled product iterations
- Use case: Consumer goods, coatings, packaging—use MLIP pre-screening to shorten cycles for stability, barrier, and mechanical property leads.
- Tools/workflows: Cloud MLIP APIs embedded in R&D PLM tools; design-of-experiments guided by MLIP + UQ.
- Dependencies/assumptions: Domain gaps (e.g., polymers/biomaterials) may require specialized or long-range-aware models; validate on representative prototypes.
Long-Term Applications
The paper outlines a trajectory toward MLIPs supplanting DFT as the primary PES engine, contingent on advances in data quality (ab initio references), long-range physics, magnetism/charge handling, task-adaptive training, and scalable Hessian computation.
Cross-Sector (Chemicals, Materials, Energy, Pharma)
- Routine DFT replacement with MLIPs trained on ab initio (e.g., CCSD(T)) data
- Outcome: Higher accuracy and lower cost than DFT for broad classes of systems; standard entry point for PES-based tasks.
- Tools/products: Next-gen foundation MLIPs with CC-level pretraining; chemistry suites defaulting to MLIP backends.
- Dependencies/assumptions: Massive, clean multi-domain ab initio datasets; scalable training; licensing/access to reference data.
- End-to-end autonomous discovery workflows (self-driving labs)
- Outcome: Closed-loop MLIP + UQ + active learning + synthesis robotics to explore materials/chemical space with minimal human intervention.
- Tools/products: Orchestration platforms integrating MLIP inference, UQ gating, automated DFT/CC fallback, and robotic synthesis/characterization.
- Dependencies/assumptions: Reliable UQ/active learning; robust prevention of catastrophic forgetting; standardized data protocols between simulation and experiment.
- Long-range and magnetic/fixed-charge-aware MLIP platforms
- Outcome: Accurate simulation of MOFs, biomacromolecules, ionic conductors, and magnetic solids in unified MLIP models.
- Tools/products: Physics-augmented architectures (e.g., MACE-POLAR-1-like) or learned all-to-all attention (AllScAIP-like) with scalable N-body approximations (e.g., fast multipole).
- Dependencies/assumptions: Efficient O(N log N) implementations; validated handling of charge/spin inputs and local magnetic moments; training data with charge/spin diversity.
- TS searches and kinetics at scale
- Outcome: Efficient Hessian-capable MLIPs enabling automated reaction path exploration and rate predictions across libraries of reactions/material transformations.
- Tools/products: MLIP variants with Hessian-efficient training/inference; reaction discovery toolkits coupling MLIP gradients/Hessians to path-finding algorithms.
- Dependencies/assumptions: Memory-efficient higher-order differentiation; curated TS/Hessian datasets; task-centric training objectives.
Software, Platforms, and Standards
- Cloud-native MLIP simulation services
- Outcome: API-first PES, forces, Hessians, and UQ as managed services with SLAs; integration into CAD/PLM and EDA tools.
- Tools/products: Serverless GPU backends; cost-aware routing (student/teacher models); compliance logging and model versioning.
- Dependencies/assumptions: Stable, versioned foundation models; governance on model drift; clear licensing/IP for model and data use.
- Continual learning with stability–plasticity guarantees
- Outcome: Foundation models that can be updated with new domains without forgetting (e.g., LoRA-like equivariant adapters, replay).
- Tools/products: Adapter marketplaces; task-conditioned routing; certified performance envelopes.
- Dependencies/assumptions: Systematic evaluation protocols; scalable replay or synthetic data generation; community standards.
- Gold-standard UQ frameworks
- Outcome: Calibrated, mathematically grounded UQ built into foundation MLIPs, validated across domains.
- Tools/products: Readout ensembles/adapters at inference; gradient- and distance-based hybrid UQ; model cards with calibration curves.
- Dependencies/assumptions: Shared benchmarks and acceptance criteria; lightweight UQ methods that scale to billion-parameter models.
Academia & Education
- Curricular shift to MLIP-first computational chemistry
- Outcome: PES instruction and research training predominantly via MLIPs, with DFT/QM emphasized for validation/data generation.
- Tools/products: Open datasets and teaching models; interactive curricula on symmetry/equivariance, UQ, and active learning.
- Dependencies/assumptions: Sustained open access to high-quality models/data; faculty training; alignment with accreditation.
- New conceptual frameworks and explainability in digital chemistry
- Outcome: Emergent concepts linking MLIP latent structure to chemical reactivity descriptors, assisting human reasoning.
- Tools/products: Latent-space analyzers, attribution tools, and interpretable surrogates.
- Dependencies/assumptions: Research on interpretability of equivariant GNNs; community acceptance of new descriptors.
Policy, Sustainability, and Economics
- Standards for ML-driven scientific evidence
- Outcome: Regulatory and funding frameworks that accept MLIP-backed predictions with documented UQ for early-stage decisions.
- Tools/products: Validation protocols; MLIP “evidence levels” analogous to experimental standards.
- Dependencies/assumptions: Demonstrated domain reliability; consensus on UQ thresholds and escalation policies.
- Large-scale reduction of HPC energy use in chemical R&D
- Outcome: Substantial cuts in compute energy by shifting routine workloads from DFT to MLIP, tracked and audited.
- Tools/products: Carbon accounting tooling integrated with schedulers; MLIP-first procurement standards.
- Dependencies/assumptions: Organizational change management; robust MLIP pipelines; ongoing monitoring.
Daily Life and Market Impact
- Faster, more sustainable product pipelines
- Outcome: Shorter time-to-market for better batteries, catalysts, and materials in consumer and industrial products.
- Tools/products: MLIP-enhanced R&D platforms; digital twins of material performance integrated into product design.
- Dependencies/assumptions: Bridging lab-to-field gaps; validation under operational conditions; IP and data governance.
- Investment and portfolio analytics for materials innovation
- Outcome: Finance and corporate strategy use MLIP-backed forecasts to prioritize R&D bets and de-risk portfolios.
- Tools/products: Decision-support dashboards combining MLIP predictions, UQ, and techno-economic models.
- Dependencies/assumptions: Transparent UQ; links to market and supply-chain models; governance around model risk.
Notes on overarching assumptions and dependencies across applications:
- Training data coverage and quality are decisive; many foundation MLIPs are trained on DFT (often PBE/PBEsol) and can inherit their biases. Movement to ab initio (CC-level) data is required for the long-term vision.
- Long-range interactions, charge states, and magnetism remain active areas; select models already incorporate these, but adoption is uneven and scaling can impact runtime.
- Uncertainty quantification must be calibrated per domain; post-hoc methods are immediately useful, but integrated, well-calibrated UQ is needed for high-stakes decisions.
- Fine-tuning risks catastrophic forgetting; favor adapter-based methods (e.g., LoRA for equivariant models) and replay strategies where generality must be preserved.
- Hardware and software stacks matter; GPUs deliver substantial speedups, and toolchains like ASE/LAMMPS with MLIP backends are the current practical path.
Glossary
- Ab initio: First-principles methods that do not rely on empirical parameters, often used as high-accuracy references in quantum chemistry. "Comparing the computational cost of ab initio methods and classical force fields with that of MLIPs is not as straightforward as one might think."
- Activation barrier: The energy threshold that must be overcome for a chemical reaction to proceed; related to reaction kinetics. "predict reaction energies and activation barriers routinely"
- Adsorption energy: The energy change when a species adheres to a surface; key for catalysis and surface science. "predicting the adsorption energies of different NH species on Ni, Ru, Ru and Ru"
- Antiferromagnetism: A magnetic ordering where neighboring spins align in opposite directions, yielding no net magnetization. "Simulating antiferromagnetism, for example, requires explicit information about local magnetic moments on individual atoms."
- Atom-centered symmetry functions (ACSF): Hand-crafted, symmetry-respecting descriptors of local atomic environments used as inputs to ML potentials. "One prominent class of such descriptors is the so-called atom-centered symmetry functions (ACSF)"
- Atomic Cluster Expansion (ACE): A systematic, symmetry-adapted expansion to model atomic environments for interatomic potentials. "the atomic cluster expansion (ACE) \cite{Drautz2019}"
- Bayesian approaches: Probabilistic methods for inference and uncertainty estimation; here, noted to have limits in DFT error quantification. "and Bayesian approaches have their limits"
- Born–Oppenheimer approximation: The separation of electronic and nuclear motion allowing electronic energies to be computed for fixed nuclei. "It is the Born-Oppenheimer approximation, the so-called clamped-nuclei approximation, which allows us to calculate the electronic energy"
- Catastrophic forgetting: Loss of previously acquired knowledge when updating a model on new data in continual learning. "this increases the risk of ``catastrophic forgetting'', i.e., a deterioration in performance in areas previously mastered"
- Charge and spin equilibration: Global adjustment of atomic charges and spins to achieve a consistent distribution under constraints. "This is followed by global charge and spin equilibration"
- Charge equilibration (Qeq): A method that assigns atomic charges by minimizing an energy functional using predicted electronegativities and hardnesses. "Other approaches use charge equilibration (Qeq) schemes"
- Charge transfer: Redistribution of electronic charge across a system, often over long distances. "This enables the model to capture long-range charge transfer and redistribution across the system"
- Clebsch–Gordan tensor product: A mathematical operation for combining angular momentum representations; used to maintain rotational equivariance. "the Clebsch-Gordan tensor product is used"
- Collinear magnetic moments: Magnetic moments constrained to align along a single axis (up or down) in a material. "which predicts collinear magnetic moments."
- Coupled cluster: A high-accuracy, systematically improvable ab initio electronic structure method. "achieve (multi-reference) coupled cluster accuracy from the outset"
- Cutoff radius: A distance threshold defining the local neighborhood considered in MLIPs. "within a cutoff radius around atom ."
- Data augmentation: Technique to enrich training data by applying symmetry-preserving or synthetic transformations. "Another option is to use data augmentation, similar to what was done in AlphaFold 3"
- Density functional theory (DFT): A quantum mechanical method that approximates electronic structure via electron density, widely used for energies and structures. "Density functional theory (DFT) has been the most important method in practice for obtaining such energies"
- Descriptor: A numerical representation of an atomic environment used as input to ML models. "This limitation has been mitigated by the introduction of descriptors, which are representations of the local atomic environment that respect the necessary symmetries"
- Dispersion: Long-range electron correlation effects (van der Waals forces) not captured by local/semi-local approximations. "semi-local DFT functionals similarly miss long-range correlations such as dispersion"
- Equivariant models: Models whose internal features transform consistently with input symmetries (e.g., rotations), enabling tensorial predictions. "This limitation is addressed by equivariant models, which are models whose features transform in the same way as the input"
- Exchange–correlation (xc) density functional: The part of DFT capturing electron exchange and correlation effects; approximated in practice. "the so-called exchange--correlation (xc) density functional"
- Fast multipole approaches: Algorithms accelerating long-range interaction calculations to better-than-quadratic complexity. "such as fast multipole approaches"
- Functional derivative: A derivative of a functional; here, the xc potential as the derivative of the xc energy functional. "as functional derivatives"
- Fukui functions: Quantities from conceptual DFT describing how electron density responds to changes in particle number; used here to enforce equilibration. "via learnable Fukui functions"
- Graph neural network: Neural architectures operating on graph-structured data, here with atoms as nodes and bonds/neighbor relations as edges. "This graph is then used as input for a graph neural network"
- Hessian: The matrix of second derivatives of energy with respect to atomic coordinates; central for vibrational analysis and transition state searches. "many applications, such as transition state (TS) searches, require Hessians."
- Knowledge distillation: Transferring knowledge from a large “teacher” model to a smaller “student” for faster inference with similar accuracy. "knowledge distillation is a promising approach."
- Kohn–Sham DFT: The standard practical formulation of DFT using single-particle orbitals; supports exchange–correlation approximations. "in Kohn--Sham DFT calculations"
- Latent Ewald Summation (LES): An approach that infers implicit charges to compute long-range electrostatics via an Ewald-like scheme without explicit labels. "An alternative to explicit charge prediction is the Latent Ewald Summation (LES) method"
- Local magnetic moments: Atom-specific magnetic moments necessary to represent magnetic ordering in materials. "requires explicit information about local magnetic moments on individual atoms."
- Long-range electrostatics: Electrostatic interactions extending beyond local neighborhoods, important for large or polar systems. "purely local MLIPs also omit long-range electrostatics."
- Low-rank adaptation (LoRA): A technique that adds trainable low-rank matrices to frozen weights to adapt large pretrained models efficiently. "called low-rank adaptation (LoRA), has been developed for LLMs"
- Message passing: Iterative exchange and update of node features along edges in graph neural networks. "a process usually called ``message passing,''"
- Moment tensor potentials: A class of ML interatomic potentials using tensorial moment descriptors. "moment tensor potentials \cite{Shapeev2016}"
- Molecular dynamics (MD): Simulation of atomic motion by integrating equations of motion using interatomic forces. "enabling stable molecular dynamics, high-throughput materials discovery, and property predictions across a vast chemical space"
- Nearsightedness of electronic matter: The principle that electronic effects are largely local, motivating locality assumptions in MLIPs. "Physically, this approach can be motivated by the ``nearsightedness of electronic matter''"
- Non-self-consistent field formalism: A scheme that updates quantities without iterating to full self-consistency, reducing cost. "using a non-self-consistent field formalism."
- PBE and PBEsol (exchange–correlation functionals): Specific generalized-gradient DFT functionals widely used in materials modeling. "errors smaller than the difference between the exchange-correlation functionals PBE and PBEsol"
- PBE0 (exchange–correlation functional): A hybrid DFT functional mixing exact exchange with PBE correlation/exchange. "the PBE0 exchange-correlation functional"
- Phonon: A quantized mode of lattice vibration; key to thermal and vibrational properties of solids. "accurately predicts harmonic phonon properties"
- Polarizability tensor: A tensor describing how a system’s dipole moment changes in response to an applied electric field. "such as dipole moments or the polarizability tensor."
- Polymorph: Different crystal structures of the same chemical composition, often with distinct properties. "the phase transition pressures of silica polymorphs under high pressure"
- Potential energy surface: A mapping from nuclear configurations to energy, underpinning reaction pathways and dynamics. "the concept of a potential energy surface"
- Semi-local DFT functional: DFT approximations depending on local quantities and their gradients, missing nonlocal correlations. "While semi-local DFT functionals similarly miss long-range correlations such as dispersion"
- Smooth overlap of atomic positions (SOAP): A descriptor based on comparing local atomic density expansions, preserving symmetries. "the smooth overlap of atomic positions (SOAP)"
- Spectral neighbor analysis potential (SNAP): An interatomic potential using bispectrum components of local environments. "the spectral neighbor analysis potential (SNAP)"
- Spherical harmonics: Functions on the sphere forming a basis for angular components; used for rotationally equivariant features. "spherical tensors represented by spherical harmonics"
- Stability–plasticity dilemma: The trade-off between retaining old knowledge (stability) and learning new information (plasticity) in continual learning. "This tension is known as the stability-plasticity dilemma"
- Transition state (TS): A saddle point on the potential energy surface corresponding to the highest-energy configuration along a reaction path. "many applications, such as transition state (TS) searches, require Hessians."
- Uncertainty quantification: Estimation of predictive uncertainty to assess reliability and guide data acquisition or model use. "rigorous uncertainty quantification is not easy"
- Wannier centres: Localized representations of electronic states used for analyzing charge distribution and polarization. "predicting partial charges or Wannier centres"
Collections
Sign up for free to add this paper to one or more collections.