- The paper introduces a multimodal foundation model that uses coarse protein-ligand representations for high-throughput binding affinity prediction.
- The methodology combines frozen pretrained encoders with a lean pairformer, achieving competitive accuracy and a 26-fold speedup over diffusion-based methods.
- Empirical results demonstrate superior Pearson correlation and calibrated uncertainty metrics, positioning TerraBind as a robust alternative in structure-based drug discovery.
TerraBind: Fast and Accurate Binding Affinity Prediction through Coarse Structural Representations
Introduction and Motivation
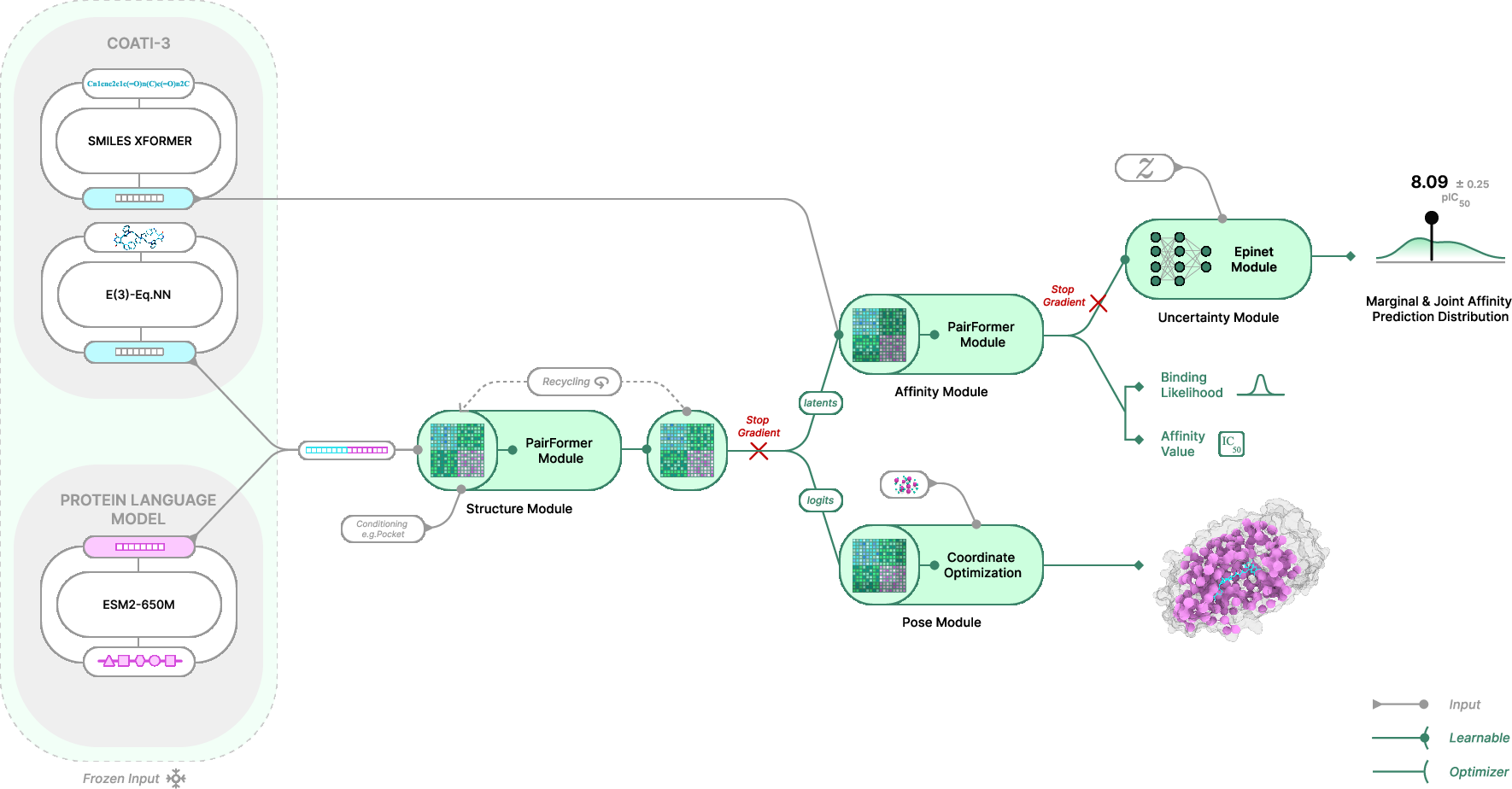
TerraBind introduces a multimodal foundation model for protein-ligand structure and binding affinity prediction that challenges the prevailing reliance on all-atom diffusion-based generative protocols in structure-based drug design (SBDD). The model’s primary hypothesis is that full all-atom resolution is unnecessary for pose and affinity prediction in small molecule drug discovery. TerraBind leverages coarse pocket-level representations—utilizing only protein Cβ atoms and ligand heavy atoms—thus radically reducing computational complexity and enabling high-throughput inference.
TerraBind’s architecture combines frozen pretrained encoders (COATI-3 for ligands and ESM-2 for proteins) and a lean pairformer trunk, sidestepping the resource-intensive diffusion processes found in methods like Boltz-2 [passaro2025boltz]. This yields a 26-fold throughput increase, making library-scale virtual screening feasible, especially in contexts such as Terray’s EMMI platform, which delivers billion-scale experimental binding datasets.
Architectural Overview
The model architecture consists of four core modules:
TerraBind’s coarse-grained approach focuses exclusively on protein residue centers (Cβ atoms, Cα for glycine) and ligand heavy atoms, streamlining geometric modeling while preserving pocket-level fidelity.
On benchmarks assessing generalization (FoldBench, PoseBusters, Runs N' Poses, proprietary crystals), TerraBind matches or surpasses diffusion-based baselines in ligand pose accuracy, demonstrating that explicit generative modeling is dispensable for the pose prediction task.
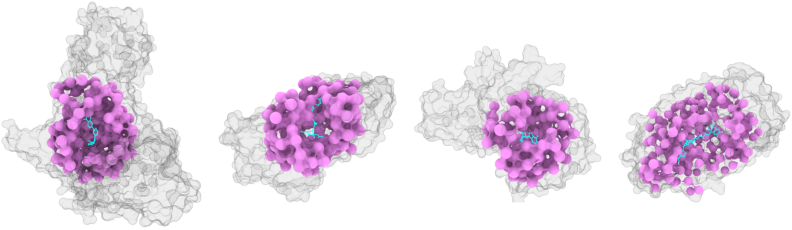
Figure 2: TerraBind coarse structure predictions for diverse protein-ligand complexes, highlighting pocket-level context without full backbone resolution.

Figure 3: Structure prediction success rates. TerraBind achieves competitive RMSD and LDDT-PLI metrics vs. full-diffusion baselines across benchmarks.
Crucially, TerraBind’s predicted ligand-protein distogram entropy tightly correlates with pose accuracy and forms a model-intrinsic confidence metric. Training data diversity is managed via a multistage curriculum encompassing experimental PDB structures and distillation from high-confidence structure predictions (e.g., AFDB, Boltz-1x) to maximize geometric generalization.
Inference Efficiency and Context Adaptation
By eliminating diffusion and operating with minimal token context centered on the binding site (typically <200 tokens), TerraBind achieves a dramatic inference speedup.
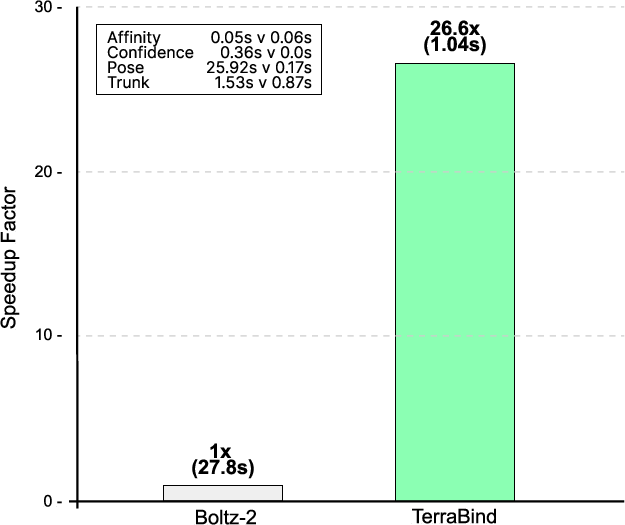
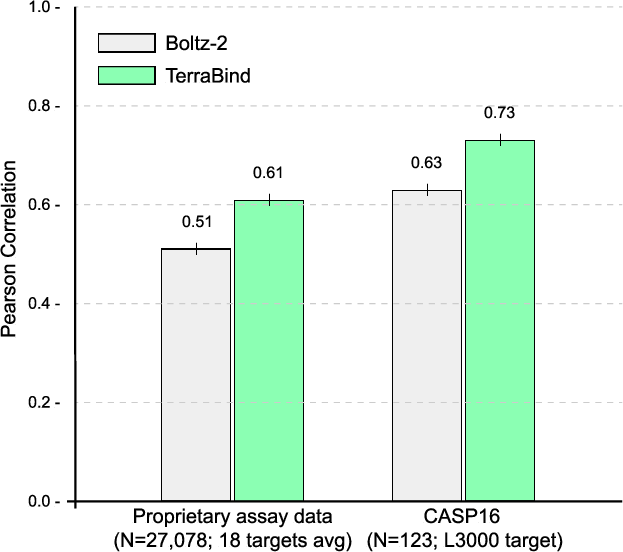
Figure 4: Inference throughput analysis. TerraBind delivers a 26× speedup per complex relative to Boltz-2, enabling high-throughput screening.
Moreover, aggressive context cropping (e.g., using predicted pocket from the largest ligand) yields a near-linear acceleration without substantial loss in affinity prediction accuracy.
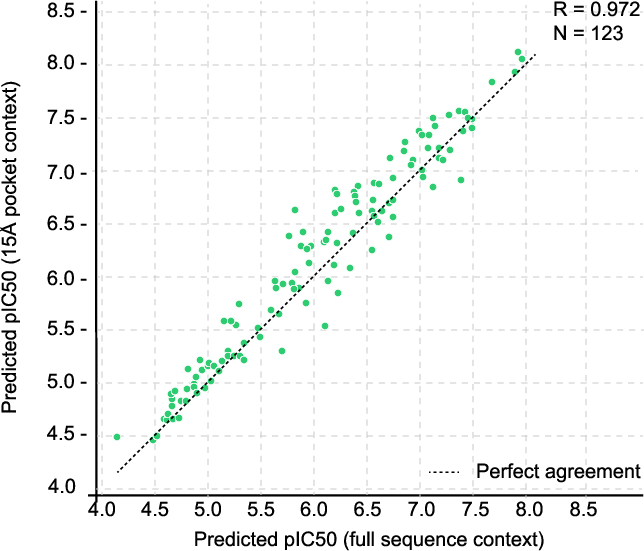
Figure 5: Minimal binding pocket context enables 17–20× speedup while maintaining 0.95 correlation with full-sequence affinity predictions.
Binding Affinity Prediction and Calibration
TerraBind’s affinity module segregates structure and affinity training via a stop-gradient protocol. Input features include pairwise structural latents, distance bin probabilities, ligand and residue embeddings. Conditioning directly on distogram bins obviates explicit coordinate input, further reducing overhead.
On CASP16 and proprietary assay data (18 targets, ≥95 IC50 measurements each), TerraBind outperforms Boltz-2 in Pearson correlation—achieving ~20% improvement and surpassing baseline on 15 of 18 benchmarks.
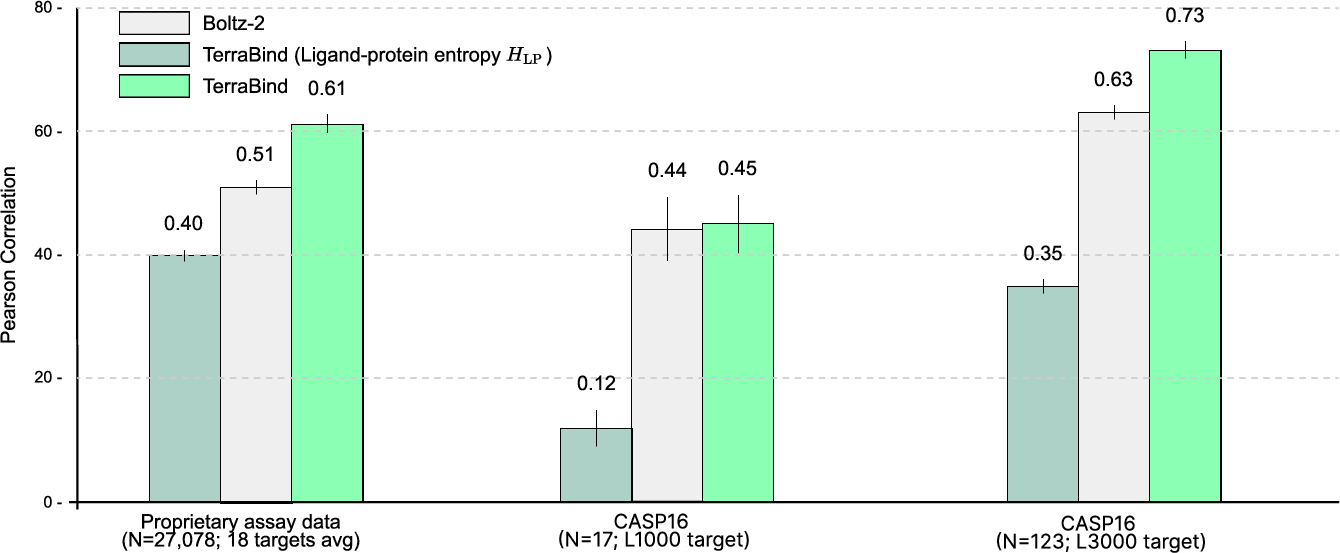
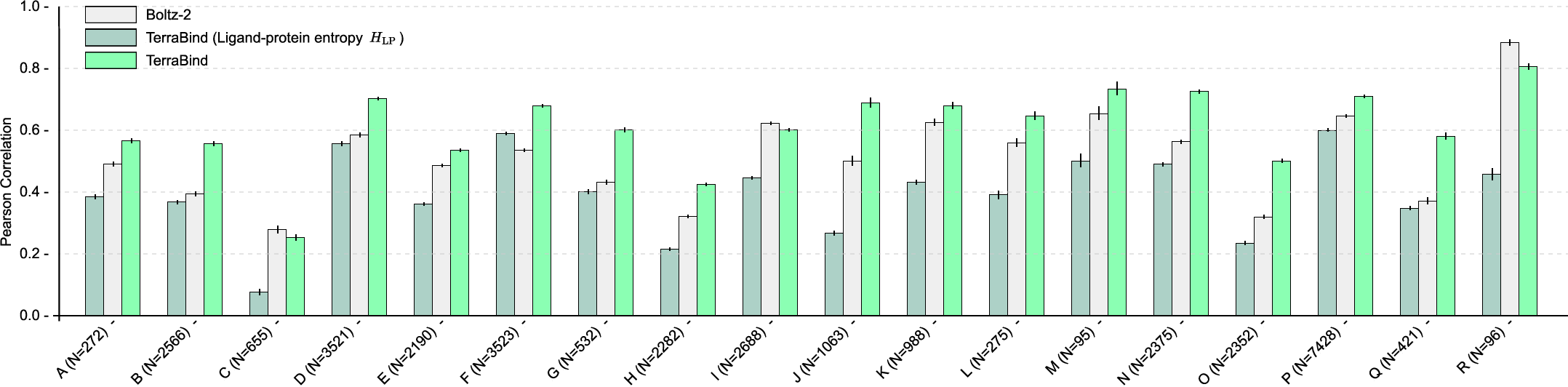
Figure 6: Pearson correlation across affinity benchmarks. TerraBind delivers consistent, target-level superiority over Boltz-2.
Structural fine-tuning on small sets of proprietary crystallographic data (<10 crystals per target) enhances affinity prediction on thousands of unseen molecules without retraining the affinity module.
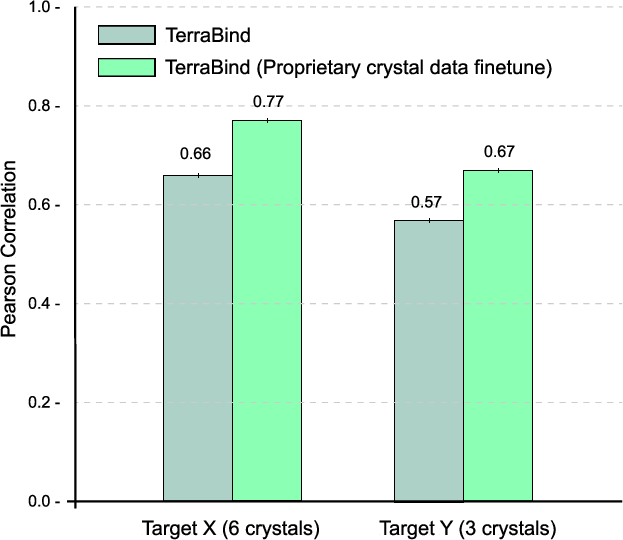
Figure 7: Structural fine-tuning on proprietary crystals boosts affinity prediction for unrelated targets using a fixed affinity module.
Uncertainty Quantification and Continual Learning
TerraBind integrates an epistemic neural network that yields calibrated affinity uncertainty, with predicted interquartile range correlating strongly with empirical accuracy.
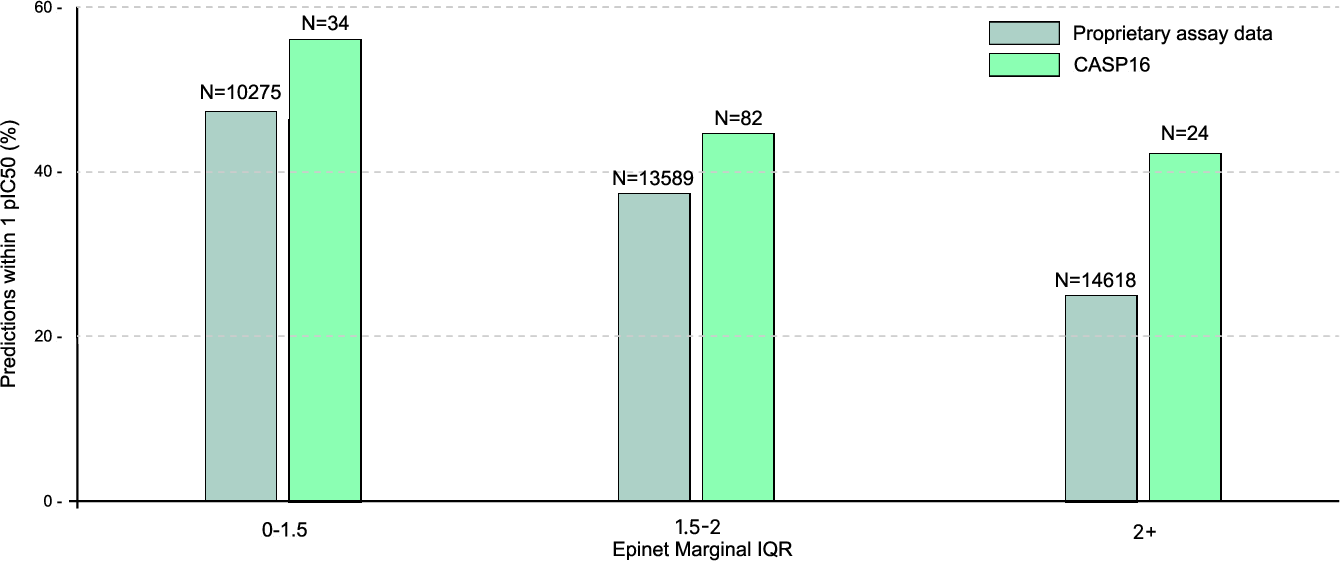
Figure 8: Epinet module calibration. Lower predicted uncertainty yields higher success rates across diverse assay datasets.
Joint epistemic distributions across compound batches unlock batch-wise hedged acquisition functions (e.g., EMAX), enabling continual learning strategies that vastly accelerate DMTA cycles and optimize hit selection. The latter exhibits a 6× improvement in IC50 affinity over traditional greedy approaches.
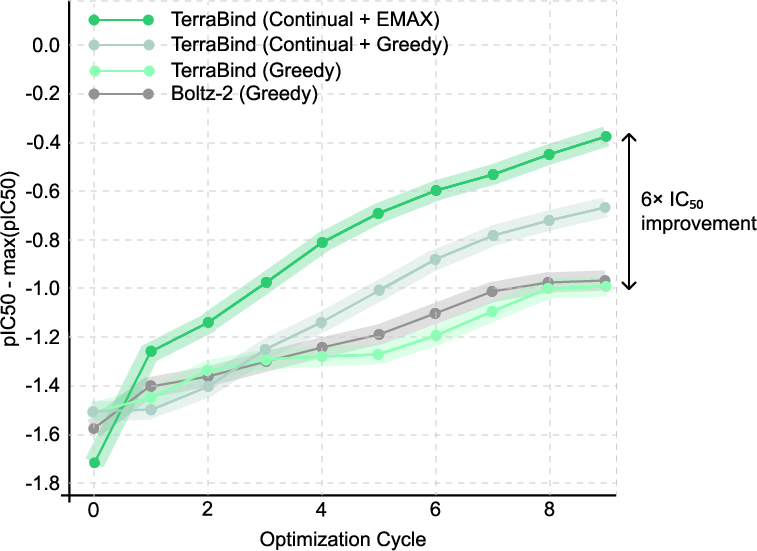
Figure 9: Continual learning with EMAX acquisition outperforms greedy strategies in simulated DMTA cycles, optimizing affinity improvement.
Practical and Theoretical Implications
- Practical Impact: TerraBind’s throughput, uncertainty calibration, and modular design make it viable for billion-scale library screening and iterative experimental design. The model can rapidly incorporate proprietary structural evidence, supporting adaptive optimization.
- Theoretical Impact: The work challenges the necessity of generative all-atom modeling for practical SBDD, emphasizing the sufficiency of coarse-grained representations for accurate affinity prediction.
- Future Directions: Expansion of chemical diversity in structural modeling (e.g., physics-based synthetic data) and refining assay context conditioning will further enhance predictive robustness. Improvements in non-Gaussian marginal modeling within the epinet module will mitigate false-positive bias and optimize batched acquisition.
Comparative Baselines
A sequence-based model with an equivalent ligand and protein encoder (MLP architecture, 5M parameters) demonstrates substantially inferior performance compared to TerraBind’s structure-aware approach, underscoring the necessity of structural representation for generalizable affinity prediction.
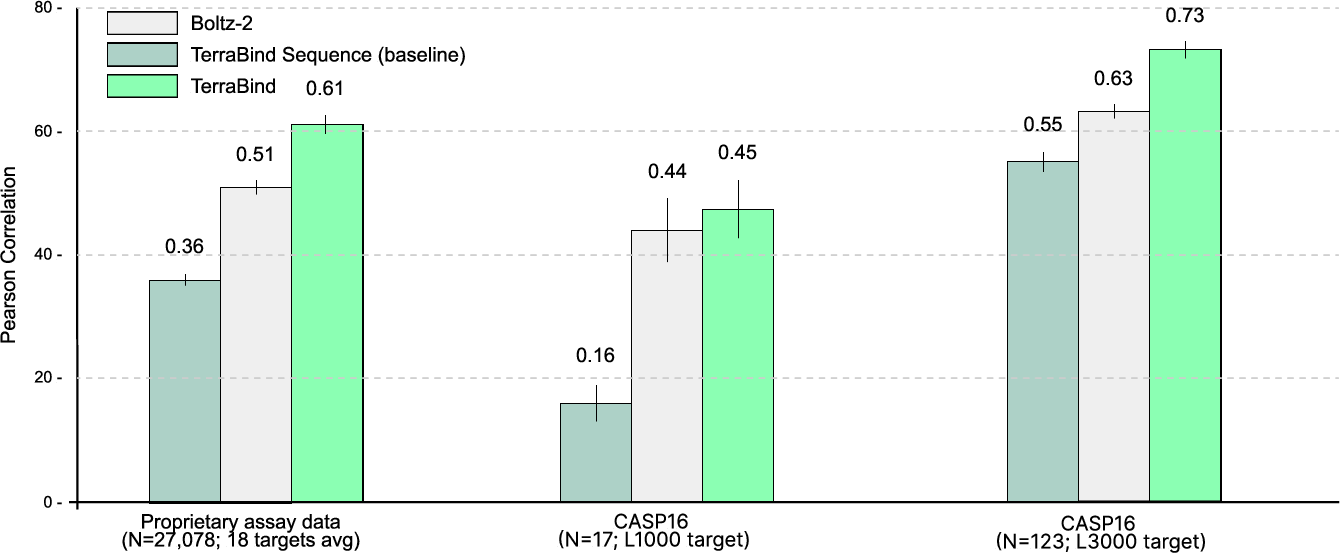
Figure 10: Sequence-only baseline performance. TerraBind achieves marked improvement by incorporating structural signal.
Limitations
- Atomistic Resolution: The architecture is suboptimal for downstream computational chemistry workflows requiring all-atom coordinates.
- Pose Generation Scalability: Optimization may not converge for large or ambiguous complexes; suitable for small molecule binding sites.
- Contextual Limitations: Narrow focus on protein-ligand interactions limits applicability in complex cell-based or mechanistic assays.
- Uncertainty Modeling: Epinet marginal distributions are prone to optimistic bias; refinement via domain-motivated prior pretraining is necessary.
Conclusion
TerraBind establishes a streamlined, modular framework for structure-based drug discovery, demonstrating that rich, coarse-grained geometric representations are sufficient for accurate pose generation and binding affinity prediction. Its high throughput, robust uncertainty calibration, and capacity for rapid fine-tuning make it practical for large-scale, iterative experimental workflows in pharmaceutical discovery. The work redefines efficiency standards in SBDD, suggesting future architectures may prioritize structural abstraction over generative atomistic modeling.
All results and architectural claims referenced herein are substantiated by the detailed analyses and visualizations presented in the original work (2602.07735).