- The paper demonstrates that minimal modifications to a standard transformer can learn molecular dynamics with state-of-the-art accuracy on QCML benchmarks.
- The MD-ET employs a triangular attention mechanism to capture three-body atomic interactions, approximating rotational equivariance through data augmentation.
- Despite lacking built-in energy conservation, the model shows promising efficiency and stability in microcanonical simulations of small molecular systems.
The paper entitled "How simple can you go? An off-the-shelf transformer approach to molecular dynamics" investigates the application of a minimally modified transformer architecture, termed the Molecular Dynamics Edge Transformer (MD-ET), to the domain of molecular dynamics (MD). The research emphasizes utilizing an "off-the-shelf" transformer with limited domain-specific adaptations, eschewing the usual inductive biases like rotational equivariance and energy conservation commonly integrated into models for MD tasks. The authors aim to assess whether such a general-purpose architecture can compete with specialized models across various molecular simulation benchmarks.
Background and Motivation
Traditional neural network approaches to MD often integrate physical inductive biases to ensure predictions adhere to known physical laws, such as rotational equivariance, where molecular forces should symmetrically respond to molecule rotations. While these biases enhance the model's data efficiency, they also complicate the architecture, limiting the model's adaptability and scalability. Meanwhile, general-purpose architectures have gained traction in other ML domains, leading to a debate on the necessity of physical constraints. This paper explores whether MD simulations can be effectively conducted using a transformer architecture with minimal MD-specific design features.
The MD-ET is an adaptation of the Edge Transformer (ET) architecture, minimally modified to suit MD tasks. Unlike specialized models, MD-ET does not enforce built-in equivariance or energy conservation. It instead employs a simple supervised pre-training approach using a large dataset of molecular structures (QCML database) to learn these properties from data.
Triangular Attention Mechanism
MD-ET utilizes a triangular attention mechanism, which performs updates on a three-dimensional tensor representation of molecules, maintaining interactions between atom pairs. This mechanism extends traditional attention by considering interactions between triples of atoms, enhancing the model's expressivity.
Experiments and Evaluation
The evaluation of MD-ET focuses on several key aspects, including accuracy, inference speed, MD stability, and generalization capabilities.
Benchmark Results
MD-ET was evaluated against current leading models on datasets such as QCML, MD17, and Ko2020:
- Performance on QCML: MD-ET achieved state-of-the-art results with a mean absolute error (MAE) lower than that of specialized MD models like SpookyNet and PaiNN.
- MD17 Benchmark: The model exhibited competitive force prediction accuracy and demonstrated remarkable efficiency in computational throughput and stability during simulations.
Equivariance and Energy Conservation
Despite lacking built-in equivariance constraints, MD-ET achieved approximately equivariant behavior through data augmentation and training strategies. Moreover, for small structures, MD-ET maintained an approximately energy-conserving behavior in microcanonical (NVE) simulations, though larger systems displayed energy drift over time.
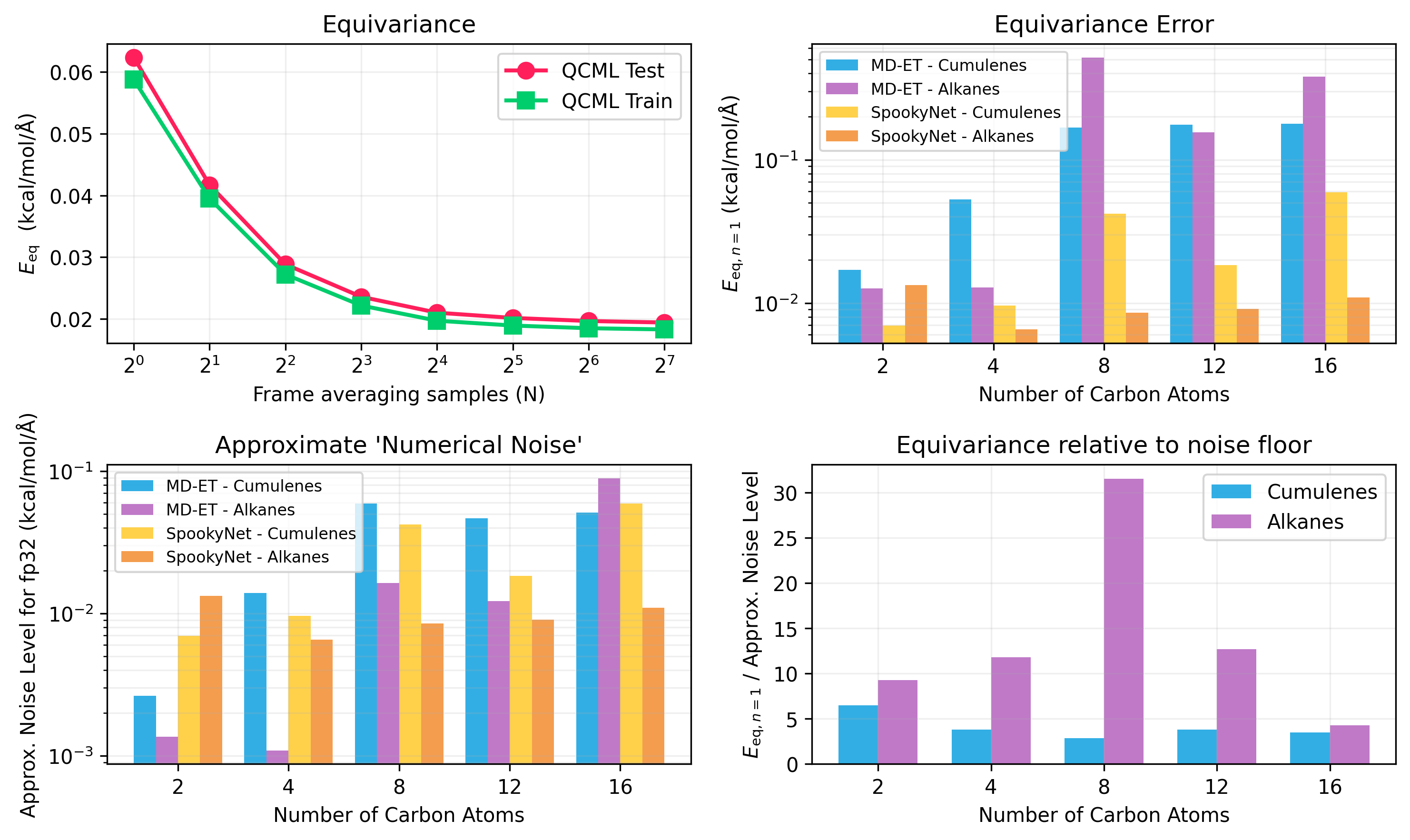
Figure 1: Equivariance Evaluation. Top Left: Average equivariance error Eeq for various frame-averaging samples. Top Right: Equivariance error over SO(3) for different molecular structures.
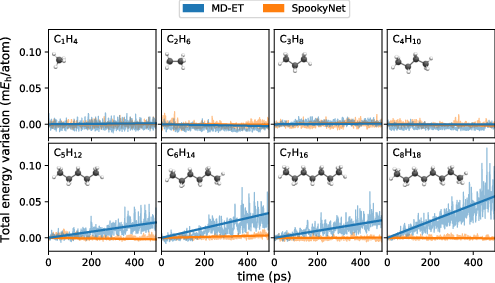
Figure 2: Total energy variation for NVE simulations of linear alkanes, comparing MD-ET and an energy-conserving model.
Real-World Applicability and Future Work
While MD-ET's current form may not be universally suitable for all MD applications, particularly larger molecular systems, it demonstrates significant potential for simpler scenarios. Its capability to learn rotational equivariance efficiently suggests that further advancements, such as incorporating additional loss terms, could enhance energy conservation learning. Moreover, MD-ET's design aligns well with current trends of scaling models to leverage larger datasets and improved hardware.
Conclusion
The paper illustrates that general-purpose transformer architectures, like MD-ET, can perform competitively in molecular dynamics without heavily relying on domain-specific inductive biases. This suggests that, with sufficient data, many of the traditionally hard-coded physics-based constraints can be learned implicitly, fostering a paradigm shift in how MD simulations might be approached with machine learning models. The research opens pathways for further exploration into the scaling potential and limitations of such unconstrained architectures in more complex molecular systems.