- The paper introduces a scalable Test-Time Training framework to enhance chemical exploration in drug design.
- It employs a modified policy-gradient RL algorithm on SMILES representations, achieving near-optimal results with 128 agents.
- The study benchmarks performance using the MolExp metric, highlighting the superiority of independent agents over cooperative RL strategies.
Test-Time Training Scaling Laws for Chemical Exploration in Drug Design
Introduction
In recent years, the application of Chemical LLMs (CLMs) using Reinforcement Learning (RL) has gained traction in the field of drug discovery. Despite their promise, these models face significant challenges, including mode collapse, which hampers their ability to explore chemical spaces efficiently. Inspired by Test-Time Training (TTT) techniques used in large-scale LLMs, this study proposes a scalable framework aimed at enhancing chemical exploration through the application of TTT to CLMs. The research introduces a novel benchmark called MolExp, designed to measure the exploration efficiency of RL-trained CLMs in discovering structurally diverse yet biologically similar compounds, reflecting realistic drug design challenges.
Methods
The study employs a modified RL approach, leveraging a policy-gradient algorithm with REINFORCE, to optimize CLMs based on SMILES representations. The temporal dynamic of chemical space exploration is modeled as a Markov Decision Process (MDP), with RL agents iteratively optimizing molecular trajectories for desired chemical properties. The novel MolExp benchmark emphasizes the identification of diverse molecules and evaluates models based on their trajectory through chemical space. This approach is further extended by introducing cooperative RL strategies, aiming to enhance the exploration efficiency through agent coordination.
Results
The experimental results reveal that scaling the TTT process by increasing the number of independent RL agents leads to a significant improvement in exploration efficiency, following a log-linear law. Figures demonstrate that with up to 128 agents, the MolExp task was nearly solved, indicating the practicality of the approach.
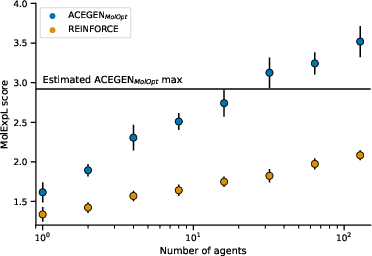
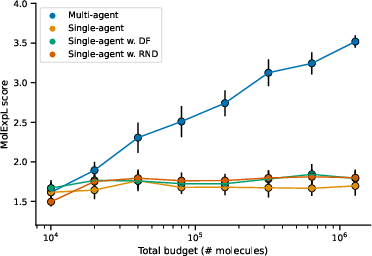
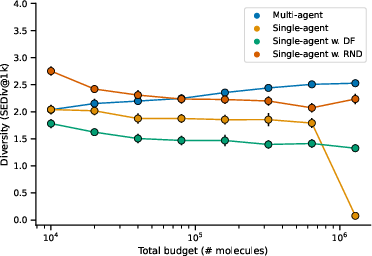
Figure 1: Scaling the number of independent RL agents, each demonstrating improved exploration efficiency on the MolExp benchmark.
Furthermore, when traditional methods of increasing TTT training time were tested, they displayed diminishing returns, highlighting the superiority of scaling in terms of agent numbers over extended training durations. Additionally, cooperative RL strategies were evaluated, and while they showed potential in enhancing diversity, they did not surpass the performance of independent agent strategies.
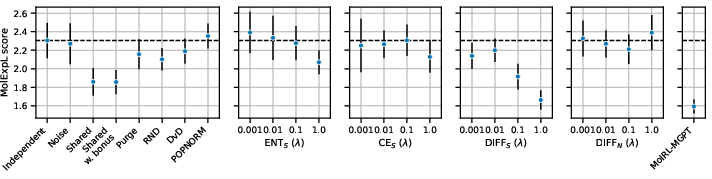
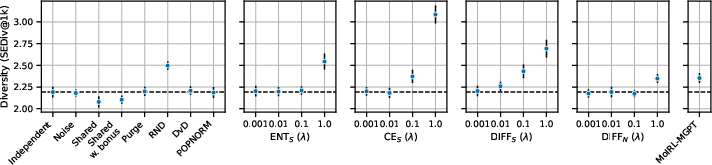
Figure 2: Performance comparison of cooperative RL strategies on the MolExpL benchmark, illustrating the challenges of achieving significant improvements over independent agents.
Implications
The proposed scaling laws for TTT present a critical advancement in the efficient exploration of vast chemical spaces, directly impacting AI-driven drug discovery processes. By optimizing agent scalability, the framework offers a clear path toward enhancing the discovery of diverse candidate molecules, ultimately accelerating and refining drug development pipelines. The MolExp benchmark sets a new standard for assessing molecular exploration capabilities, emphasizing the need for diverse and efficient chemical exploration.
Conclusion
This study provides a novel framework for scaling TTT in CLMs to improve chemical exploration efficiency significantly. The introduction of the MolExp benchmark underscores the importance of efficient search strategies in drug discovery and sets the stage for future research focused on optimizing and diversifying algorithmic approaches for generative molecular design. As computational strategies continue to evolve, the scalability and adaptability of RL approaches in CLM contexts will be pivotal in overcoming current limitations in AI-driven drug discovery.