- The paper introduces a novel CNN-based framework that reformulates gene expression data into a matrix for accurate human age estimation.
- It employs a unique data augmentation method with random Gaussian noise to overcome small dataset challenges and improve model performance.
- Experimental results reveal a notable MAE of 3.69 and MdAE of 4.1, demonstrating the framework’s superiority over previous methodologies.
Human Age Estimation from Gene Expression Data using Artificial Neural Networks
Introduction
The use of gene expression data for human age estimation presents an intriguing opportunity to advance our understanding of the biological aging process, with profound implications for fields such as healthcare and forensic science. The work "Human Age Estimation from Gene Expression Data using Artificial Neural Networks" introduces a novel framework that leverages both raw and augmented gene expression data from human dermal fibroblasts to improve age estimation accuracy. This paper addresses challenges associated with data representation, limited sample sizes, and harnesses neural networks to facilitate this estimation process, showcasing the framework's superiority over existing methodologies.
Data Representation and Augmentation
The research posits a two-dimensional spatial representation for gene expression data, reformulating traditional vectorized data into a matrix format. This transformation aims to enhance the ability to exploit non-adjacent gene relationships through the use of CNNs, a strength of the spatial data layout devised by the authors.

Figure 1: Spatial data representation for three individuals; left to right: 1 year old, 30 years old and 61 years old.
Additionally, the model overcomes the limitation of the small dataset size via data augmentation. A novel approach generates new synthetic data without significantly altering the statistical data distribution by incorporating random Gaussian noise. This method allows the model to generalize better when applied to unseen data, as demonstrated by the improved accuracy metrics.
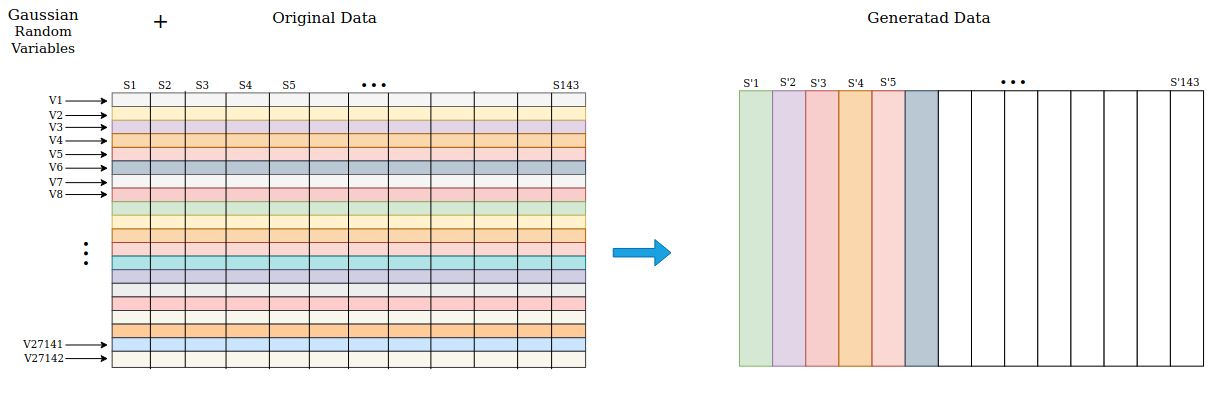
Figure 2: Data Augmentation Scheme.
Neural Network Framework
The framework is centered on a shallow neural network designed specifically to handle the spatial, augmented gene datasets for age estimation. The architecture relies on a series of convolutional layers supplemented by max-pooling operations and activation functions, optimized for the unique characteristics of the gene expression data matrix.
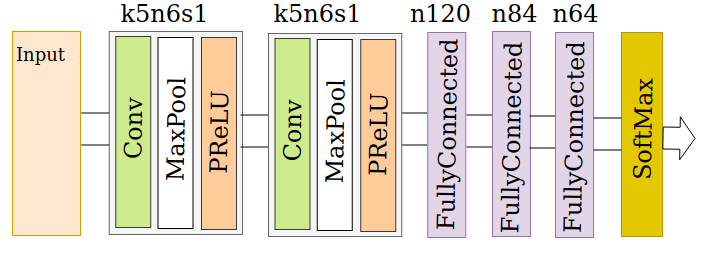
Figure 3: Network architecture for age estimation via an ensemble of age-group classifiers.
The models classify individual samples into predetermined age groups amalgamated into a comprehensive age estimation using an ensemble approach, ensuring robustness despite the dataset's inherent limitations. The innovation in framework design expands the capacity to extract meaningful insights from what could ostensibly be sparse data on its own.
Experimental Results
Empirical results validate the proposed framework's efficacy, demonstrating superior performance in terms of mean absolute error (MAE) and median absolute error (MdAE) compared to leading earlier methodologies.
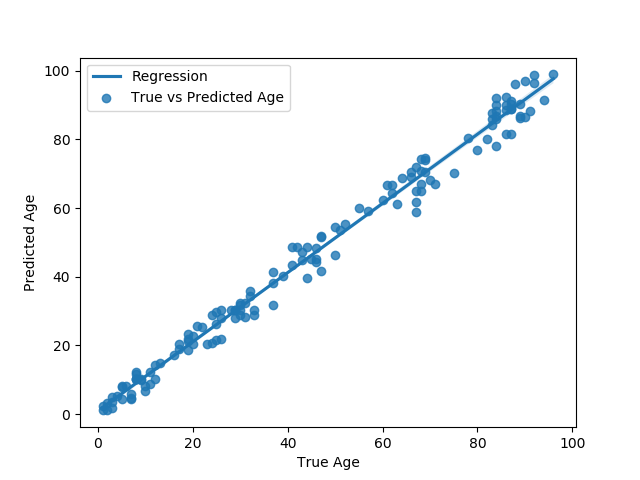
Figure 4: True vs predicted age.
Key findings from the experiments indicate the effectiveness of data augmentation and novel representation combined with a purpose-built CNN, achieving a notable MAE of 3.69 and MdAE of 4.1. These results underscore the potential for leveraging this framework across similar genomic estimation tasks, marking a significant improvement over previous studies.
Conclusion
The framework outlined in "Human Age Estimation from Gene Expression Data using Artificial Neural Networks" highlights the benefits of innovative data representation and augmentation strategies alongside tailored neural network architectures in deriving precise age estimations from genetic data. Such advancements hold promise for substantial improvements in personalized medicine applications and forensics. Future research could further refine this method and expand its applicability across other genomic datasets, enhancing the robustness and accuracy of phenotype predictions.

