- The paper presents an information-theoretic framework that identifies potential age-related genes in human dermal fibroblast data.
- It employs both unsupervised and semi-supervised learning techniques, utilizing metrics like KL divergence and cosine similarity.
- The approach refines gene clusters to identify 82 novel age-related genes and highlights age 40 as a critical threshold.
The paper "An Information-Theoretic Framework for Identifying Age-Related Genes Using Human Dermal Fibroblast Transcriptome Data" (2111.02595) presents a sophisticated methodology for identifying genes associated with aging using human dermal fibroblast transcriptome data. By leveraging machine learning techniques and information-theoretic principles, the authors aim to elucidate hidden patterns in gene expression that correlate with the aging process.
Proposed Framework
The proposed framework involves a multi-step process consisting of unsupervised and semi-supervised learning techniques to analyze gene expression data. Starting with a dataset of 27,142 genes, the approach utilizes clustering and supervised refinement to identify potential age-related genes.
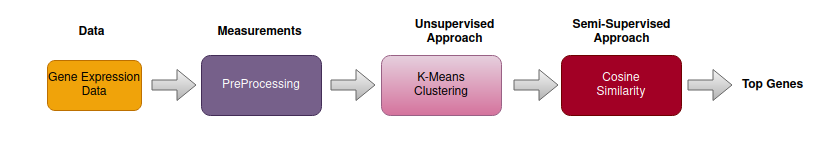
Figure 1: Proposed pipeline for identifying age-related genes.
Unsupervised Learning Approach:
In the initial phase, the framework employs unsupervised learning techniques to detect key features using information-theoretic measures such as entropy, Kullback-Leibler (KL) divergence, and correlation scores. These features are utilized to cluster gene expression data into groups that reveal associations with aging. The binary clustering strategy aims to classify genes based on their age-related activity patterns.
Feature Ranking:
The authors apply information-theoretic measures to rank genes based on their potential age relevance. By computing these metrics for different age thresholds, the study identifies significant features and selects genes with the highest scores for downstream analysis.
Semi-Supervised Refinement
In the subsequent phase, the framework refines its selection of age-related genes using semi-supervised learning. This involves applying knowledge from previously identified genes known for age association. Cosine similarity and Jensen-Shannon divergence are used as similarity measures between newly identified genes and known age-related genes. The semi-supervised process refines clusters, yielding curated gene sets that are further sub-clustered using k-means clustering to isolate specific gene functionalities.
Evaluation and Results
The authors rigorously evaluate the framework's effectiveness by contrast against a dataset of 307 known age-related genes. The use of KL divergence emerged as a crucial element for recognizing these genes, with the unsupervised phase successfully isolating the majority of known genes. The framework's semi-supervised learning phase enhanced gene selection, ultimately identifying 82 novel genes previously not associated with aging.
The study identifies age 40 as a critical threshold that demarcates significant changes in gene expression patterns related to aging. This finding is consistent with other research suggesting that the midlife period marks a key physiological transition.
Discussion and Conclusion
This study demonstrates the potential of integrating machine learning with information-theoretic principles for biological data analysis. By focusing on human dermal fibroblast transcriptome data, the researchers provide insights into age-related changes in gene expression. The results suggest that information theory can be a valuable tool in gene expression analysis, specifically for unveiling age-related genetic markers.
Moreover, the methodology has implications for developing therapeutic strategies to modulate aging processes and extend healthspan based on gene expression modulation. Future developments may focus on enhancing the computational framework with more extensive datasets to improve the robustness and accuracy of gene selection.
By employing a comprehensive methodological approach, this paper offers an effective strategy for discovering age-related genes, contributing to our understanding of the aging process and laying groundwork for future bioinformatics research.

