- The paper presents a novel de novo molecular structure elucidation pipeline that bypasses the need for ground-truth molecular formulas by leveraging mass-shell constraints.
- It integrates a self-supervised neural encoder for rapid fingerprint prediction with a Transformer-based block-diffusion decoder, ensuring candidates meet experimental mass constraints.
- Experimental evaluations demonstrate superior Top-1 accuracy and structural quality, highlighting MARLIN's potential in untargeted metabolomics and related fields.
Introduction and Motivation
The identification of small molecules from tandem mass spectrometry (MS/MS) data remains a significant bottleneck within metabolomics, natural products, and exposomics. The vast majority of spectra acquired in untargeted experiments correspond to compounds not present in existing libraries, limiting the scope of traditional database search approaches. While state-of-the-art de novo molecular structure elucidation methods exist, nearly all assume access to the correct molecular formula—a requirement that is impractical in true discovery settings, given the unreliability of formula prediction itself.
MARLIN addresses this core limitation by entirely eliminating dependence on ground-truth molecular formulas at any stage. Instead, it directly elucidates molecular structure from spectral data, leveraging a self-supervised neural encoder for rapid fingerprint prediction and a block-diffusion LLM decoder operating under a mass-shell constraint. This constraint ensures that proposed structures are consistent with the measured experimental precursor mass, providing a principled and reliable alternative to formula conditioning.
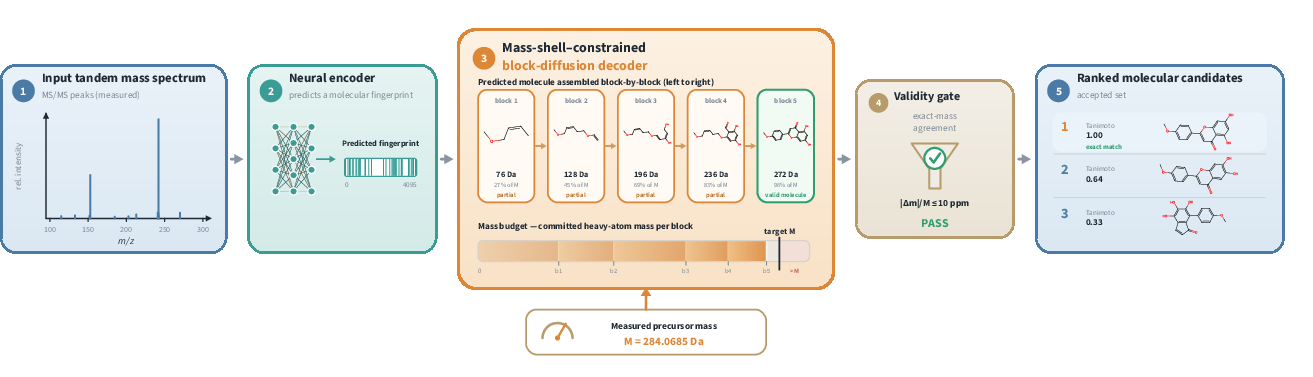
Figure 1: Overview of the MARLIN spectrum-to-structure pipeline, showing direct fingerprint prediction from spectrum, mass-shell constrained block-diffusion decoding, and validity gating—entirely without molecular formula computation.
Methodology
Central to MARLIN is the neural encoder that predicts a high-dimensional molecular fingerprint from the raw spectrum. The pipeline utilizes the DreaMS encoder, a self-supervised model trained on large-scale spectral–structure pairs, which operates solely on peak m/z and intensity pairs, requiring neither explicit formula nor candidate enumeration. The resulting 4096-bit Morgan fingerprint prediction is used for subsequent conditional generation, with optional ablation to alternate encoders such as MIST.
Mass-Shell Constrained Block-Diffusion Decoding
Molecular structure generation proceeds via a Transformer-based block-diffusion LLM. Unlike prior approaches which fix the atom inventory using a formula, MARLIN's decoder leverages the experimentally measured precursor mass M as both its sole stoichiometric condition and as a dynamic constraint on generation.
- Conditioning: The decoder is conditioned on the predicted fingerprint, precursor mass (Fourier-feature-encoded), and isotope pattern.
- Block-wise Masked Diffusion: Decoding occurs in contiguous token blocks. At each step, the decoder commits to tokens corresponding to molecule sub-units, frozen once finalized.
- Mass-Shell Prune: At each unmasking step, the decoder masks out any token selection that, if committed, would cause the cumulative heavy-atom mass to exceed M (plus tolerance), as calculated from precomputed token masses. This prune is proven never to exclude the true molecular structure, as it operates only on dead-ends provably inconsistent with the observed mass.
- Hydrogen-aware Termination: The decoder estimates maximal achievable hydrogen count based on valence budget to prevent premature termination or mass budget overshoot, coupling end-of-sequence with mass reachability.
- Validity Gating: Structural candidates are parsed post each block. Only valid chemical structures within specified mass error (ppm) tolerance are accepted.
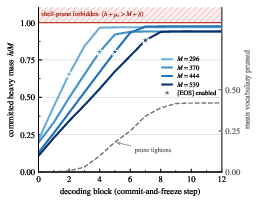
Figure 2: Mass-shell constrained decoding: the cumulative committed mass rises with each block, rarely exceeding M due to pruning, with the hydrogen contribution resolved at the valence-aware acceptance step.
Robust Candidate Diversity
Block-diffusion decoders typically collapse to a single candidate when re-sampled. To ensure diversity, MARLIN independently perturbs the fingerprint conditioning for each candidate, using random dropout. This process yields a rankable set of valid structures for each spectrum, each satisfying the stringent mass constraint.
Symmetric Fingerprint Noise Objective
Recognizing inevitable errors in fingerprint prediction, MARLIN introduces symmetric corruption during decoder training: both random dropping and addition of fingerprint bits. This approach trains the decoder for robustness to real-world encoder errors, improving reliability under noisy conditions.
Experimental Results
MARLIN is evaluated on the NPLIB1 benchmark, focusing on spectrometry-based de novo structure generation without ground-truth formula information.
- Top-1 Accuracy: MARLIN achieves 16.94% using the formula-free DreaMS encoder (and 19.18% with MIST as encoder), outperforming all prior formula-unknown de novo methods and, notably, surpassing even formula-privileged methods such as MBGen and DiffMS under this more realistic constraint.
- Structural Quality: Average Top-1 Morgan Tanimoto similarity reaches 0.55, the highest among non-formula methods, indicating consistently near-native predictions even when exact structures are not recovered.
- Mass-Shell Validity: Mass-shell constraint ensures every candidate matches the measured mass within tolerance, effectively recovering the correct formula as a byproduct in 76.7% of cases, nearly matching dedicated predictors.
Ablation studies demonstrate that candidate diversity and mass conditioning are crucial for accuracy and coverage. Removing the fingerprint perturbation drops Top-1 accuracy by 4% (to 12.83%), and omitting mass conditioning similarly degrades performance. Notably, adding conditioning diversity to a full-sequence diffusion baseline (FRIGID) does not confer similar benefit, underscoring the synergy of block-diffusion and conditioning perturbation in MARLIN.
Qualitative assessments confirm the system's error mode: near-misses are highly fingerprint-similar structural isomers, primarily indistinguishable by MS/MS due to inherent ambiguity, with differences in regio- or stereochemistry rather than core scaffold.
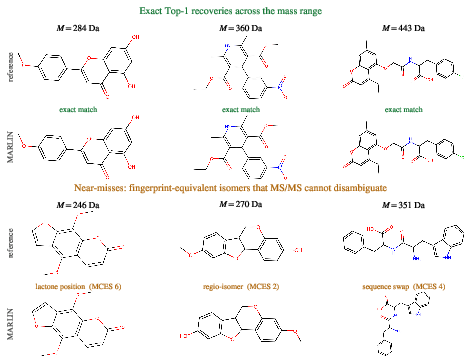
Figure 3: Representative example outputs—exact Top-1 structure recoveries (top) and archetypal near-misses (bottom), highlighting typical cases where MARLIN's candidate deviates from the reference primarily in classical, MS/MS-indistinguishable isomerism.
Implications and Future Directions
By discarding reliance on formula prediction and operating entirely within an observable-induced mass constraint, MARLIN fundamentally shifts the operational regime for de novo spectral elucidation to the setting observed in real-world discovery pipelines. In practical terms, this enables robust, scalable annotation of previously uncharacterized molecular species, facilitating advances in untargeted metabolomics, drug discovery, environmental screening, and biomarker identification.
The theoretical contributions—particularly the formulation and proof of mass-shell-safe decoding—offer a new paradigm for constrained generation tasks where strict global constraints must be satisfied without explicit specification. The practical impact is compounded by a significant reduction in computational cost and error propagation compared to formula-dependent pipelines.
Future research will likely extend MARLIN's decoder to accommodate larger molecules (addressing the combinatorial complexity and hydrogen budget expansion), integrate multi-modal spectral features, or further improve fingerprint encoding through larger-scale self-supervised training. Advances in fingerprint encoder architectures, extension to multitask or multi-instrument settings, and scaling to fragmentation spectra from even more diverse chemical space remain open and important directions.
Conclusion
MARLIN establishes a new standard in de novo molecular structure elucidation from tandem mass spectra by removing dependence on ground-truth molecular formulas. Its mass-shell constrained block-diffusion architecture, robust to encoder error and able to generate diverse and valid candidates, demonstrates clear superiority over prior works in the no-formula setting on challenging benchmarks. The implications are both practical and theoretical, enabling direct, high-confidence elucidation of novel chemistries previously inaccessible by traditional methods, and offering a generalizable framework for constraint-aware generative modeling in chemistry and beyond.

