- The paper demonstrates that SAP-X2C systematically reduces two-electron picture-change errors and outperforms SNSO-X2C in key EPR, NMR, and X-ray absorption measurements.
- SAP-X2C extends the X2C framework to property and response calculations, accurately reproducing a range of spectroscopic parameters for heavy-element systems.
- The approach offers a non-empirical correction with computational cost parity, providing a robust and transferable alternative for modeling relativistic effects.
SAP-X2C for Spectroscopy: Comparative Analysis with Screened Nuclear Spin-Orbit Approximations
Overview and Theoretical Foundation
This paper analyzes the application of the superposition of atomic potentials exact two-component (SAP-X2C) methodology to molecular spectroscopy, specifically benchmarking it against established screened nuclear spin-orbit X2C (SNSO-X2C) corrections. The work extends SAP-X2C from its established ground-state energy domain to property and response calculations, including NMR, EPR, Mössbauer, UV/vis, and X-ray absorption spectra. The theoretical differentiation between SAP-X2C and SNSO-X2C centers on how each approaches the two-electron picture-change error (2ePCE) arising in one-electron X2C (1e-X2C) Hamiltonians.
In SAP-X2C, an atomic-potential-based correction is introduced directly to the one-electron potential and its relativistic extension within the X2C decoupling transformation, designed to capture both scalar-relativistic and spin-orbit 2ePCE contributions. This approach is non-empirical and yields a well-defined thermochemical limit. Contrastingly, SNSO-X2C employs empirically fitted rescaled spin-orbit terms, with various parameter sets (Boettger, mSNSO, SNSO-DC etc.), treating only the spin-orbit part of 2ePCE and leaving the scalar component uncorrected other than via error cancellation.
Assessment of Spectroscopic Properties
EPR Hyperfine Coupling and g-Tensor
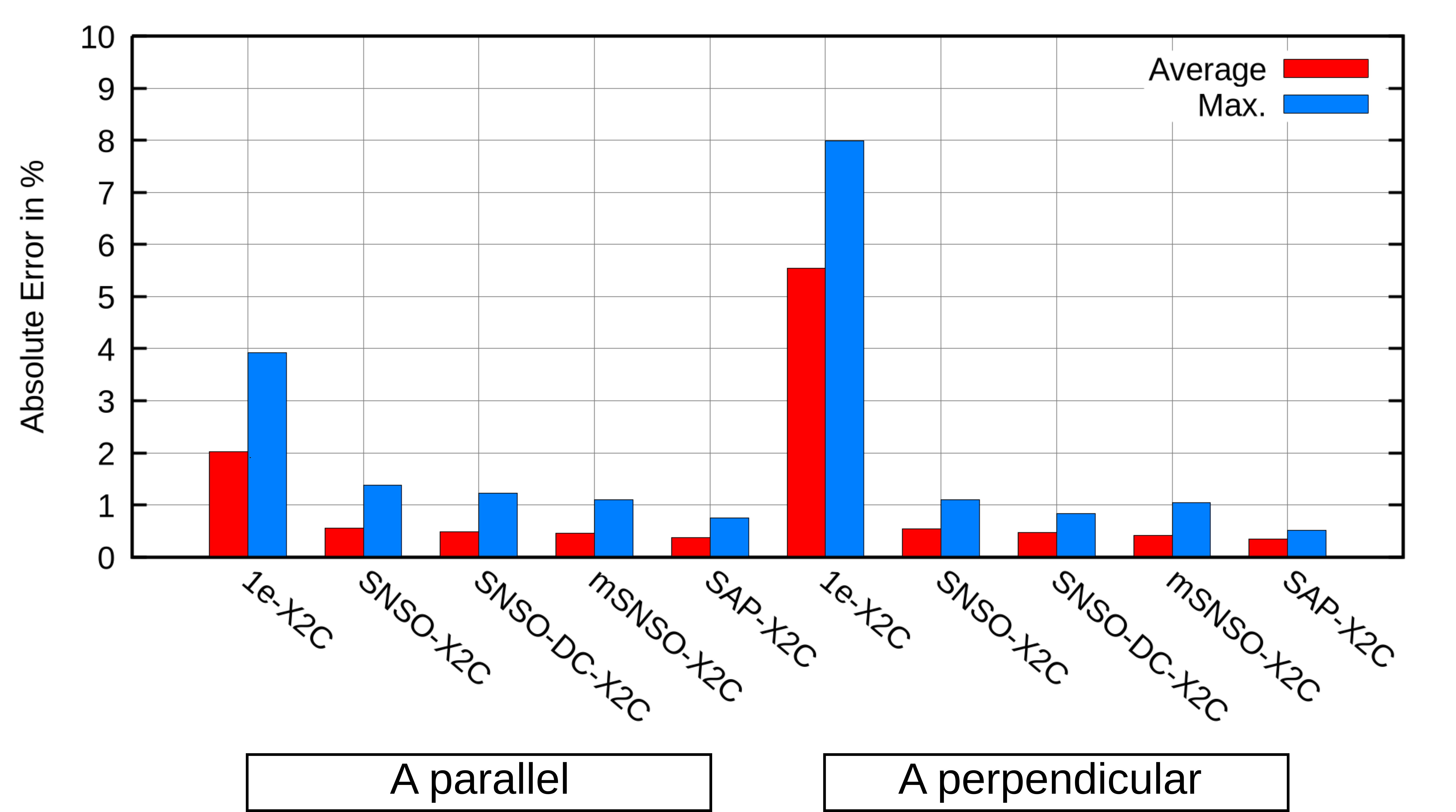
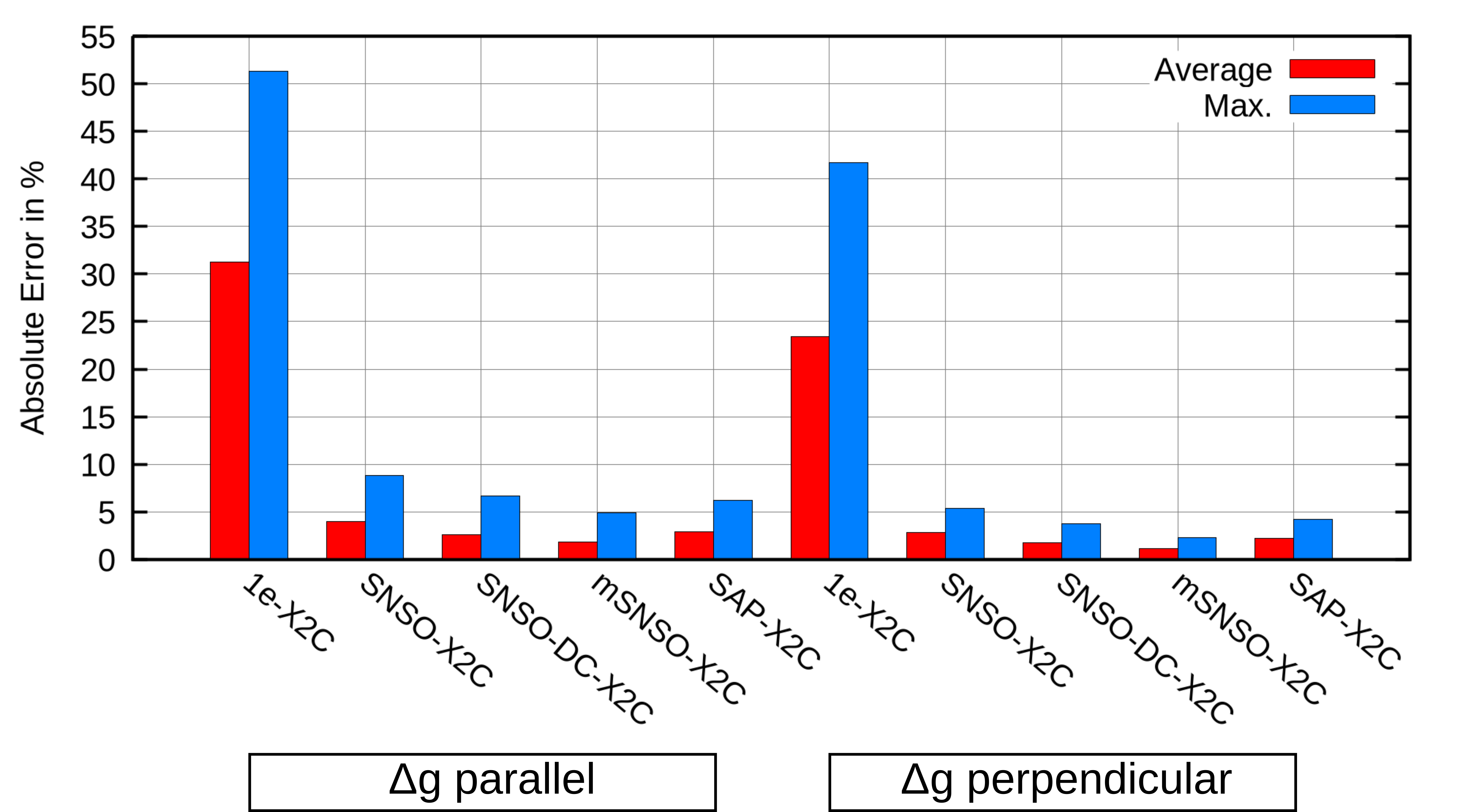
The hyperfine coupling tensor and principal components of g-shift were evaluated for SAP-X2C and several SNSO-X2C parameterizations across a canonical test set of 17 transition-metal complexes. Both mean and maximum errors with respect to four-component (4c) Dirac-Coulomb references were analyzed.
For the EPR hyperfine couplings:
Regarding the g-shift:
Atomic and molecular NMR shielding constants, particularly for heavy elements and their hydrides, show that:
- SAP-X2C outperforms all SNSO-X2C approaches for molecules, especially for systems with high spin-orbit coupling, though mSNSO remains competitive due to specific parameterization (notably optimized for Xe).
- For atomic shielding, mSNSO is typically slightly better than SAP-X2C for noble gases, consistent with its parameterization, but SAP-X2C’s more ab initio character confers greater transferability.
- Both relative and absolute contact densities in Mössbauer spectroscopy are well reproduced by SAP-X2C; SAP-X2C is less dependent on error cancellation than SNSO, thus provides a more robust framework for absolute properties.
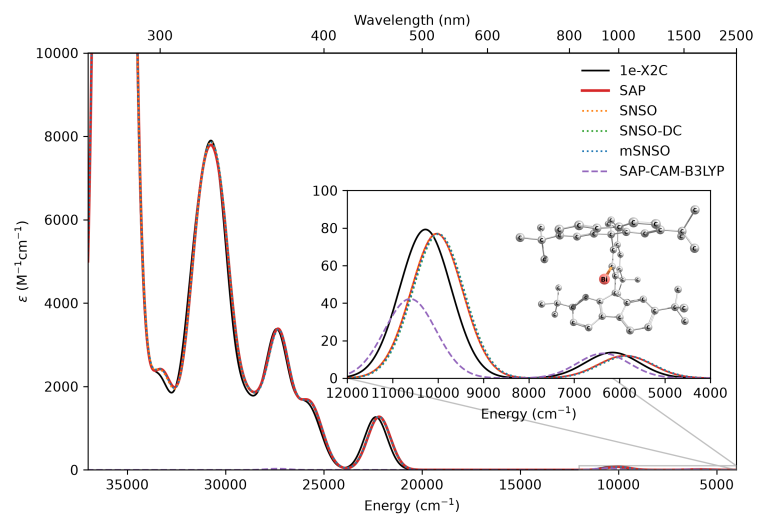
UV/Vis Spectroscopy and Zero-Field Splitting
Study of a hydrindacene bismuthinidene complex revealed that:
X-ray Absorption Spectroscopy: Core-Level Edges
X-ray absorption edge energies (K and L edges) are highly sensitive to relativistic picture-change effects:
- Both SAP-X2C and optimized SNSO-X2C yield significant error reductions compared to 1e-X2C, halving mean absolute deviations.
- SAP-X2C gives the most accurate individual edge positions in comparison to 4c-DKS and atomic mean-field (amfX2C/ eamfX2C) approaches, especially for elements with pronounced 2ePCE.
- For chemically meaningful properties (e.g., spin-orbit splitting), all 2ePCE-corrected methods converge within 0.5 eV but differences are observed at the level of individual edge positions and depend on the chosen reference.
Practical and Theoretical Implications
The SAP-X2C methodology represents a non-empirical, transferable, and rigorously defined protocol for correcting two-electron picture-change errors for a wide suite of properties. Strong numerical evidence, especially the systematic reduction below 1% error for EPR parameters and sub-10 ppm for heavy-atom NMR shieldings, supports the claim that SAP-X2C is preferable over SNSO approaches for property calculations in the X2C framework. Importantly, SAP-X2C is also less reliant on error cancellation for accurate relative quantities, supporting its extension to absolute property calculations and spectroscopy of heavy-element systems.
The formal simplicity and computational cost parity with SNSO approaches, combined with public availability of required basis sets and straightforward integration into standard quantum chemical codes, argues for SAP-X2C to supplant empirical 2ePCE corrections as the default for advanced spectroscopic simulations.
The residual advantages of atomic mean-field corrections (amfX2C, eamfX2C) are restricted to high-level correlated wavefunction treatments or applications demanding the absolute minima in 2ePCE, at a cost of significant practical complexity.
Future Directions
The comprehensive generalization of SAP-X2C to response theory, as presented in this study, opens the way for systematic improvements in the accurate prediction of relativistic effects in molecular spectroscopy. There are several avenues meriting further exploration:
- Optimization of SAP basis sets for property-specific X2C calculations could potentially surpass even the best current empirical SNSO variants.
- Extension to correlated wavefunction methods, including coupled-cluster and multi-reference treatments, should be assessed regarding both accuracy and efficiency.
- Integration of SAP-X2C corrections into high-throughput computational spectroscopic workflows and their validation across further unexplored regions of the periodic table.
Conclusion
This study substantiates SAP-X2C as an efficient, systematically improvable approach for the correction of two-electron picture-change errors in spectroscopic property calculations using the X2C framework. SAP-X2C yields errors that are equal to or smaller than those of widely used SNSO-X2C corrections, and it eliminates the need for empirical parameterization. Its formal rigor and implementation simplicity strongly motivate its adoption as the preferred standard for relativistic property calculations in molecules containing heavy elements (2607.03814).