- The paper introduces Gr-SA, a cache-aware Graph Suffix Array that drastically reduces I/O operations for pattern matching on deterministic Wheeler pangenome graphs.
- It leverages heavy-light decomposition and unary path decomposition to achieve sub-microsecond query latencies and up to 500x speed improvements compared to traditional methods.
- Empirical results validate near-optimal I/O complexity and practical memory usage, setting a new standard for scalable genomic data analysis.
Cache-Efficient Pattern Matching on Deterministic Wheeler Pangenome Graphs
Introduction
The paper "Faster Cache-Efficient Pattern Matching for Deterministic Wheeler Pangenome Graphs" (2607.02113) analyzes the limitations of existing Burrows-Wheeler Transform (BWT)-based pattern matching algorithms on Wheeler automata, especially in the context of large pangenomic graphs. While the extension of BWT principles to automata (Wheeler deterministic finite automata, or WDFAs) allows for efficient pattern queries, the original backward/forward search approach triggers I/O costs proportional to the pattern length, which restricts practical scalability for large datasets typically seen in bioinformatics applications. The work presents a novel, cache-aware indexing technique, termed the Graph Suffix Array (Gr-SA), that drastically reduces cache misses at the expense of increased—but manageable—memory footprint.
Methodology and Data Structures
The authors generalize the classic string suffix array paradigm to deterministic Wheeler automata by exploiting the total order on states induced by Wheeler automata properties. The core operation, determining the occurrence of a pattern P in a WDFA with n states, is decomposed into three phases:
- Prefix Localization: Identify the longest prefix β of P such that β matches at least two distinct states, leveraging infimum and supremum strings associated with WDFA states. This search operates in O((Bm+logn)lognlogm) I/O operations, with m=∣P∣ and cache block size B.
- Transition Verification: If P=βaγ, efficiently probe for the existence of a transition labeled a out of states matched by n0, using succinct signature representations that enable n1 I/O probes, with n2 the alphabet size.
- Path Navigation: For the pattern suffix n3, traverse the automaton using path-decomposed data (unary path decomposition) to ensure the number of cache misses is bounded by n4, with n5 being the number of maximal unary paths traversed.
A salient component is the use of heavy-light decomposition on infimum and supremum pseudoforests, ensuring path queries in the automaton can be executed cache-efficiently.
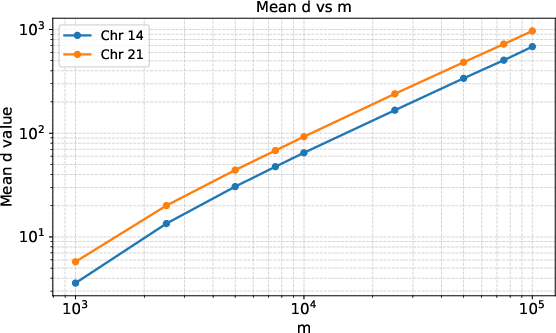
Figure 1: Mean number of traversed unary paths n6 during locate(n7) as function of pattern length n8 (log-log scale), averaged over n9 random queries on pangenome graphs. β0 is typically two orders of magnitude less than β1.
Empirical Evaluation
The Gr-SA approach was benchmarked on Wheeler pangenome graphs derived from the dbSNP database, specifically on human chromosomes 21 and 14, and compared to a baseline forward search BWT index. Empirical results confirm that for realistic genomic graphs, the parameter β2 (number of unary paths traversed) is about two orders of magnitude smaller than the pattern length β3 (as seen in Figure 1), confirming the practical cache efficiency of the approach.
Key results include:
- Speed: The Gr-SA index is up to 500x faster than traditional forward search, with per-character query latencies as low as β4 ns versus nearly β5s for the baseline.
- Space: The Gr-SA consumes up to 15x more space than the forward search index, but remains within practical limits—never exceeding β6 GB RAM even on the largest graphs tested.
- Cache Miss Profile: The improved index drastically reduces random access operations by interleaving binary search over automaton state orders with large contiguous memory scans enabled by the unary path decomposition.
Theoretical Analysis
The paper supplies rigorous proofs showing that the Gr-SA index has, under realistic assumptions about automaton sparsity (as present in biological pangenome graphs), near-optimal I/O complexity for subpath queries. By partitioning the computation over β7, each phase's complexity is controlled, and infimum/supremum forests, path coherence, and input-consistency properties guarantee correctness and state interval contiguity.
For patterns with multiple occurrences, the index achieves improved time and space complexity, returning all matched states without explicit traversal, requiring only a single binary search operation.
Implications and Future Directions
The results presented strongly suggest that suffix array-inspired, cache-efficient pattern matching strategies can scale to the demands of gigascale pangenomic data, provided automaton determinism and input-consistency properties (e.g., Wheeler order) are satisfied. The techniques may generalize to non-genomic workloads involving large, sparse, regular languages—potentially impacting search, verification, and string processing tasks in both theory and computational practice.
Two divergent directions suggest themselves for future research:
- Compressed Representations: Addressing the memory blowup, perhaps via succinct or compressed representations of the unary path map and transition tables, may bring Gr-SA to even larger graphs—e.g., population-scale human pan-genomes.
- Extensions to Nondeterminism or Partial Wheeler Orders: The extension of these results to Wheeler NFAs or partial Wheeler graphs, common in certain bioinformatics contexts, remains open and nontrivial.
Conclusion
"Faster Cache-Efficient Pattern Matching for Deterministic Wheeler Pangenome Graphs" (2607.02113) rigorously demonstrates that indexing strategies inspired by suffix arrays and heavy-path decompositions overcome the primary I/O bottleneck of BWT-pattern matching on large Wheeler automata. By judiciously trading moderate additional RAM usage for drastic I/O reductions, the proposed Graph Suffix Array allows sub-microsecond queries on gigascale pangenome graphs, with robust empirical validation supporting the strong theoretical claims. This work sets a new bar for scalable, cache-aware pattern matching in the context of deterministic automata, especially relevant to population genomics and large-scale sequence analysis.