- The paper introduces a HCAO model extending anharmonic oscillator methods to polyatomic species in DSMC.
- It employs local mode analysis with Morse potentials to capture bond-specific anharmonicity and rapid vibrational energy redistribution.
- Simulations for H2O, HCN, and CH4 validate the model against traditional TCE benchmarks, enhancing predictive accuracy in non-equilibrium conditions.
Harmonically-Coupled-Anharmonic-Oscillator Model for Polyatomic Chemistry in DSMC
Introduction and Motivation
High-enthalpy gas flows, such as those encountered during atmospheric entry processes, push established thermochemical models to their limits due to the pronounced non-equilibrium and complex intramolecular energy dynamics of polyatomic species. Conventional Direct Simulation Monte Carlo (DSMC) methods often employ the assumption of uncoupled harmonic vibrators for internal molecular modes, ignoring anharmonic effects as well as couplings that become significant at elevated excitation and in the proximity of dissociation and reactive events. The challenge becomes acute when modeling polyatomic species, where normal mode analyses fail to accurately reflect bond-specific excitations and energy redistribution.
To enable more accurate simulations under such non-equilibrium conditions, this paper extends the anharmonic oscillator model—which was previously limited to diatomic molecules [Civrais et al. 2023]—to polyatomic species using a harmonically-coupled-anharmonic-oscillator (HCAO) framework. The new model is tailored for DSMC applications and is integrated into the PICLas code base with explicit treatment of anharmonicity and local-bond-specific intramolecular redistribution.
Theoretical Model: From Harmonic Approximations to HCAO
Potential Energy Surfaces and Limitations of Normal Modes
The vibrational structure and reactivity of molecules is determined by their potential energy surface (PES). For diatomics, a 1D PES can be well described by the Morse potential, which captures both vibrational anharmonicity and bond breaking, unlike the classical harmonic oscillator approximation which fails for highly excited states or during bond dissociation.
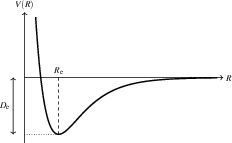
Figure 1: Potential energy curve for a diatomic molecule as a function of bond length, also illustrating the equilibrium bond distance Re and well depth De.
Polyatomic molecules possess high-dimensional PESs leading to $3N-6$ vibrational degrees of freedom (for non-linear species). Standard normal mode analyses, based on diagonalization of the Hessian at a PES stationary point, yield collective delocalized vibrations. This approach inadequately describes bond-localized excitations and reactive events, especially for highly excited vibrational states and systems far from equilibrium.
Local mode analysis, instead, employs internal coordinates corresponding to specific bonds, producing vibrations localized on diatomic and triatomic fragments. Bond-specific anharmonicity is characterized in local coordinates, while remaining bending vibrations are often well-approximated as harmonically-coupled normal modes.
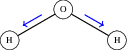
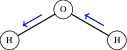
Figure 2: Illustration of a symmetric stretch vibrational mode in water, representing a normal mode.


Figure 3: OH1 stretch in water as a local mode, with vibrational excitation localized to a single bond.
Kinetic Rate Theory with HCAO
Chemical reactivity in gases is fundamentally controlled by the distribution of molecular energy among vibrational and translational degrees of freedom. RRKM theory extends transition state theory to unimolecular gas-phase reactions, formalizing the connection between the energy state of a molecule and its reaction rate. Intramolecular vibrational redistribution (IVR) channels energy into reactive coordinates. While harmonic models leave energy localized, real systems with anharmonic couplings allow efficient energy flow—a precondition for accurately modeling dissociation and reaction rates.
The HCAO approach models local stretching as independent Morse oscillators (explicitly treating anharmonicity), harmonically coupled via bending modes. This coupling supports rapid redistribution of vibrational energy and introduces mode-specific access to reaction coordinates, thus enabling a physically grounded link between intramolecular dynamics and reactivity.

Figure 4: Energy profile along a reaction coordinate with activation energy Ea and reaction enthalpy ΔH.
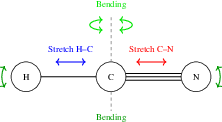
Figure 5: The HCN molecule in the local mode system with two stretching modes and two bending modes.
Implementation in DSMC: Algorithmic Details
The model is integrated into PICLas, augmenting the treatment of internal molecular energy assignment and reactive event sampling. All local stretching modes utilize Morse potentials parameterized by ab initio or spectroscopic data. Bending modes are retained as harmonic oscillators. Vibrational state populations are sampled using discrete energy levels weighted according to Boltzmann statistics.
At each collision event, collisional excess energy relative to the dissociation or reaction threshold is calculated—including careful subtraction of zero-point energies per the explicit local mode basis. The stochastic acceptance-rejection method ensures the correct quantum population sampling after relaxation and reaction events.
Crucially, for bimolecular exchange reactions, only a fraction αj of the vibrational energy of each mode may couple to the reaction coordinate, in line with statistical models and trajectory studies (e.g., SVP model, trajectory calculations for CH4 + H [Jordan & Gilbert 1995]). Steric and energetic selectivity is enforced through probabilistic models for orientation and energy disposal.
Numerical Verification: Comparison to Existing Models
Several representative systems are simulated in closed-cell DSMC configurations to validate the HCAO-based model against the traditional Arrhenius-based TCE model. The simulations assess both unimolecular and bimolecular reactions as well as recombination-dissociation equilibria across a range of temperatures and molecular species.
Water Dissociation
Simulations of H2O dissociation in presence of N2 as a non-reactive collider reveal that the HCAO-based approach matches both reaction rates and product energy distributions obtained with the Arrhenius-based TCE model for both particle number evolution and OH product temperature.
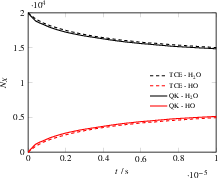
Figure 6: Temporal evolution of simulation particle numbers for HDe0O dissociation, showing agreement between QK+HCAO and TCE models.
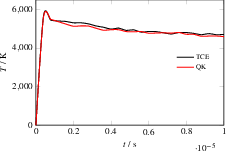
Figure 7: OH temperature evolution during water dissociation, comparing QK+HCAO and TCE approaches.
HCN Dissociation
The system is probed at both 5000 K and 12000 K to test the model's ability to capture temperature-dependent branching among multiple dissociation channels (HCN De1 H + CN and HCN De2 HC + N). The HCAO model accurately tracks the TCE reference at lower temperatures. At higher temperatures, it additionally captures secondary channel participation and the limitations of Arrhenius fits outside calibrated temperature regimes, highlighting a distinct advantage for non-empirical modeling.
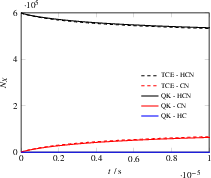
Figure 8: Simulation particle number evolution for HCN dissociation at 5000 K, comparing QK+HCAO and TCE.
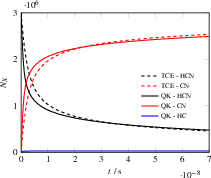
Figure 9: Simulation particle number evolution for HCN dissociation at 12000 K, highlighting additional channel recognition by QK+HCAO.
Methane: Exchange and Equilibrium
For the hydrogen-exchange reaction (CHDe3 + H De4 CHDe5 + HDe6), the model correctly implements mode-specific coupling to the reaction coordinate and reproduces reference kinetics. The model's capability is further validated in the non-trivial equilibrium of CHDe7 dissociation and recombination, where identical kinetics and equilibrium concentrations emerge across both model classes.
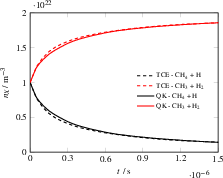
Figure 10: Species number densities over time for the CHDe8 + H De9 CH$3N-6$0 + H$3N-6$1 exchange reaction using HCAO and TCE models.
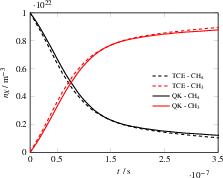
Figure 11: Number densities during the recombination-dissociation equilibrium of methane, QK+HCAO vs. TCE model.
Implications and Outlook
The HCAO-based extension enables first-principles modeling of vibrational anharmonicity and energy redistribution effects in polyatomic reaction dynamics within DSMC. This approach removes the dependence on empirical global kinetic fits, directly linking simulation-level reactivity to underlying ab initio or spectroscopic vibrational parameters and reaction barriers. The method offers a seamless transition from fundamental molecular physics to macroscopic flow simulation—especially critical for high-temperature, high-excitation phenomena in aerospace applications.
Potential future directions include:
- Explicit incorporation of additional anharmonic corrections in recombination rates.
- Validation and adjustment via non-invasive measurements or state-resolved experiments in extreme environments (e.g., shock tunnels, atmospheric re-entry).
- Extension to state-resolved reaction modeling, allowing for richer non-equilibrium descriptions as per recent state-to-state relaxation studies [Kustova et al.].
- Integration of rotational-vibrational couplings using projections from ab initio trajectory analyses.
Conclusion
The harmonically-coupled-anharmonic-oscillator (HCAO) model presented provides an advanced, physically motivated platform for polyatomic chemistry modeling in DSMC. By systematically embedding anharmonicity and local-mode-specific couplings, it delivers predictive accuracy for reaction rates and product energy distributions across a variety of prototypical molecular systems. This advance strengthens the theoretical and computational foundation for non-equilibrium chemical simulations, positioning DSMC for broader application in high-enthalpy chemistry and kinetic theory.

