Fast genomic read alignment with minibwa
Abstract: Motivation: BWA-MEM remains a popular short-read mapper especially for the purpose of variant calling. Several groups have accelerated this algorithm as it has been the performance bottleneck of many current workflows. However, constrained by the original design, these drop-in replacements could only achieve limited speedup. Breaking changes to BWA-MEM are required for further improvement. Results: We developed minibwa for aligning short and accurate long reads against a reference genome. It combines BWA-MEM variable-length seeding with minimap2 chaining and base alignment. It speeds up BWA-MEM2 further with additional prefetch for seeding, new heuristics to skip unnecessary mate rescue and reduced effort in highly repetitive regions where reads would anyway be wrongly mapped due to structural changes. Minibwa is about four times as fast as BWA-MEM and over twice as fast as BWA-MEM2 at comparable accuracy. It also natively supports directional bisulfite sequencing data to high mapping accuracy. Availability and implementation: https://github.com/lh3/minibwa
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What this paper is about
This paper introduces a new computer program called “minibwa.” Its job is to quickly and accurately figure out where short pieces of DNA (called “reads”) belong on a known reference genome, like matching tiny puzzle pieces back into a huge picture. This step, called “read alignment,” is a basic part of many genetics tasks, such as finding variants (differences in DNA) or measuring DNA methylation (a chemical tag on DNA).
Minibwa is designed to be much faster than older, popular tools (especially BWA-MEM) while keeping the same high accuracy. It also works well for both short reads (from common DNA sequencers) and accurate long reads (from newer technologies). Plus, it natively supports bisulfite sequencing, a special method used to study DNA methylation.
What questions did the researchers ask?
In simple terms, the team wanted to know:
- Can we make DNA read alignment much faster than BWA-MEM without losing accuracy?
- Can we handle both short reads and accurate long reads with one tool?
- Can we avoid wasting time in very repetitive DNA regions where reads are hard to place anyway?
- Can we build in strong support for bisulfite sequencing data (used in methylation studies)?
- Can we reduce the cost and time of common genome analysis pipelines, like variant calling?
How did they do it? (Methods explained with analogies)
Think of the genome as a gigantic book with billions of letters (A, C, G, T), and each DNA “read” as a sentence fragment you want to place back into the right spot in that book.
Here are the main ideas, with everyday analogies:
- A compressed index (FM-index/BWT): This is like a compact, smart index for the giant book so you can quickly find where a small phrase appears. Minibwa keeps BWA-MEM’s efficient index format but uses it more cleverly to save time.
- Seeds and SMEMs (super-maximal exact matches): Instead of comparing whole sentences right away, you first look for exact word chunks (seeds) that match the book. SMEMs are the longest exact chunks that are not contained in any larger chunk. Minibwa finds these seeds faster and in batches, like checking many phrases at once instead of one-by-one.
- Chaining (connecting anchors): Once you’ve found several exact chunks that match nearby spots in the book, you connect them like linking puzzle pieces to outline where the read most likely fits. Minibwa borrows a strong chaining method from another tool (minimap2), which is great for both short and long reads.
- Alignment (fine-tuning the match): After the chunks are connected, you still need to compare letter-by-letter to handle small differences (like typos or small gaps). Minibwa starts with a quick “no-gaps” check and only runs the heavier, detailed comparison (dynamic programming) if needed. It also uses SIMD, a hardware feature that lets the computer compare many letters in parallel—like scanning several lines at once.
- Memory prefetching and batching: Getting data from main memory is slow (like walking to a warehouse), but getting it from the CPU’s cache is fast (like grabbing from a nearby shelf). Minibwa “calls ahead” (prefetches) and batches many lookups so the data is ready in the fast shelf by the time it needs it. This keeps the CPU busy and speeds things up a lot.
- Smarter behavior in repetitive regions: Some genome parts (like centromeres) are extremely repetitive—many different places look almost the same—so short reads can’t be placed confidently. Minibwa avoids overspending time there, since those reads can’t be trusted anyway.
- Paired-end rescue with a quick filter: For pairs of reads, if one is placed and the other is missing, the program tries to “rescue” its partner nearby. Minibwa uses a fast rough check (counting short word matches) before doing expensive alignment, saving time by skipping bad candidates.
- Bisulfite sequencing support (for methylation): Bisulfite sequencing changes certain C letters to T in a predictable way. Minibwa builds a special index that expects these changes and uses scoring that treats C-to-T differences as allowed, but T-to-C as suspicious. This makes methylation mapping both fast and accurate.
What did they find? (Main results and why they matter)
In tests on real and simulated data, minibwa:
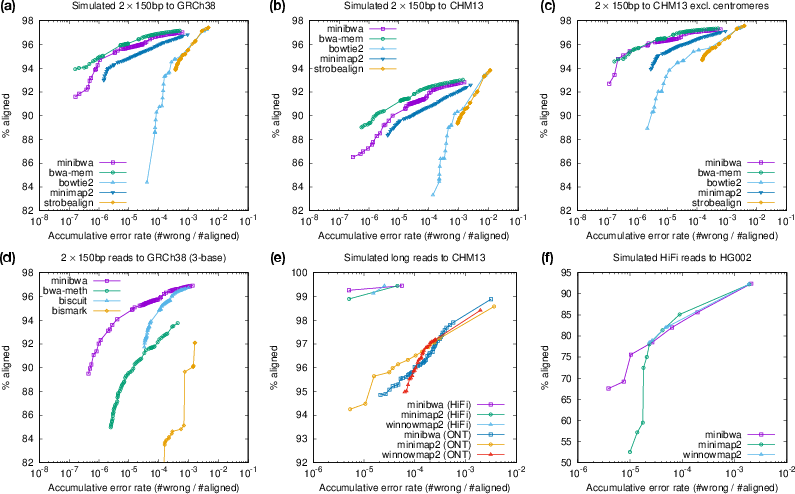
- Runs about 4× faster than BWA-MEM and over 2× faster than BWA-MEM2 on common short-read whole-genome data, with similar accuracy.
- Matches or slightly improves downstream variant calling accuracy (the end goal for many users), compared to BWA-MEM.
- Works well on accurate long reads, performing similarly in speed to minimap2 (and much faster than some long-read-only tools).
- Maps bisulfite sequencing data faster and with high accuracy, simplifying methylation studies.
- Uses less than 20 GB of memory in the tested scenarios, which is modest for whole-genome alignment.
Why this matters: Read alignment is a major time and cost bottleneck in genome analysis. Making it 2–4× faster without losing accuracy can save labs and clinics a lot of computing time and money and speed up research and diagnostics.
What’s the impact?
- One tool for more jobs: Minibwa handles short reads, accurate long reads, and bisulfite reads, so teams can simplify their pipelines.
- Faster pipelines: Variant calling and methylation analysis can run much faster at scale, making it easier to process many genomes.
- Practical accuracy: Even though minibwa spends less time in tricky repetitive regions, it maintains high accuracy where it counts—like in variant calls that clinicians and researchers use.
- Open and evolving: It’s open-source, actively developed by authors known for BWA-MEM and minimap2. The team plans to improve support for special reference features in the future.
In short, minibwa is a faster, modernized DNA read aligner that keeps accuracy high, works across different sequencing technologies, and makes big genomic analyses more efficient.
Knowledge Gaps
Unresolved Gaps, Limitations, and Open Questions
Below is a concise list of concrete gaps and open questions that remain after this work; each item highlights a specific area future researchers could address.
- Missing support for alternate contigs and pangenome/graph references
- The tool does not support alt-contigs (explicitly acknowledged) or graph/pangenome mapping, which are increasingly important for diverse populations and SV-rich regions; a redesigned, validated alt/graph-aware mapping mode is needed.
- Limited evaluation beyond small variant calling on short reads
- Only DeepVariant SNP/indel calling was assessed; no evaluation for CNV/SV detection, read phasing, allele-specific analyses, or coverage uniformity, especially in complex genomic regions.
- Long-read accuracy assessed only on simulated data
- Real HiFi/ONT accuracy (including in segmental duplications, SD-rich loci, and CMRG/medically relevant genes) is not benchmarked; evaluate against curated truth sets and SV callsets on real data.
- Reduced effort in highly repetitive regions lacks downstream impact analysis
- The approach de-prioritizes centromeres/acrocentrics, lowering theoretical accuracy there; quantify trade-offs for analyses that rely on repeat mappings (e.g., centromere/acrocentric CNV, repeat instability, telomere/centromere studies) and consider user-tunable modes.
- Mapping quality (MAPQ) calibration is unvalidated
- No assessment of MAPQ calibration across read types (short/long/BS-seq/Hi-C) and genomic contexts; provide calibration curves and miscalibration diagnostics to ensure MAPQ reflects empirical error rates.
- Bisulfite sequencing: scope and accuracy not fully characterized
- Only directional BS-seq is supported; non-directional, oxidative bisulfite, and EM-seq are not. There is no evaluation of methylation calling accuracy (CpG/CH contexts), strand bias, or alignment correctness on BS-seq-specific simulations/real truth datasets.
- Hi-C/chimeric read handling not specialized
- Although “supports Hi-C”, there is no dedicated logic for chimeric ligation junctions, no split-read modeling tailored to Hi-C, and no evaluation of contact map artifacts or MAPQ reliability for inter/intra-chromosomal pairs.
- Parameterization is heuristic without sensitivity analyses
- Key thresholds (seed length 19, second-round s ≤ 10, up to 50 chains, mate-rescue q-mer filter M_t ≥ 10) and the read-length adaptation function are not systematically optimized or validated across error profiles, read lengths, and platforms.
- Formula for length-adaptive parameters appears incomplete and unvalidated
- The provided expression for θ(ℓ) is syntactically incomplete in the manuscript and lacks empirical validation; a complete specification and ablation showing robustness across ℓ distributions is needed.
- SIMD and architecture-specific optimization space underexplored
- DP uses SSE4.1/NEON; AVX2 showed no gain and AVX-512/SVE were not examined. Analyze bottlenecks (compute vs memory bandwidth), explore wider vectors, tiling strategies, and NUMA effects; provide scaling on diverse microarchitectures.
- Multi-thread scaling and cache contention not characterized
- Only 32-thread performance is reported; provide strong scaling/efficiency curves, analyze prefetch interactions under high thread counts, and assess NUMA-aware scheduling.
- Indexing time, disk space, and memory–performance trade-offs not reported
- Especially for BS-seq (4× reference versions), quantify index build time/size, RAM vs locate sampling rate r, and precomputation (e.g., 10-mer tables); provide modes for low-memory environments.
- Generalizability to non-human and polyploid/high-repeat genomes untested
- No benchmarks on large/complex plant genomes, polyploid species, or microbial metagenomes; evaluate accuracy, speed, and memory in these contexts and tune defaults accordingly.
- Handling of ambiguous bases (N) may introduce artifacts
- Reference Ns are converted to random bases; quantify impacts near N-blocks on mapping errors and false confidence, and consider N-aware scoring or masked seeding.
- Base-quality usage in scoring not detailed
- It is unclear whether mismatch penalties are base-quality aware (as in BWA-MEM); assess the effect of quality-aware vs fixed scoring on variant calling accuracy, especially for error-prone reads.
- Reporting policy for secondary/supplementary alignments and SAM tags not documented
- Downstream tools rely on SA/MC/AS/XS/MQ tags and split alignment conventions; specify and validate compatibility with common pipelines (duplicate marking, GATK, SV callers).
- Lack of ablation studies on speed contributors
- No breakdown of how much prefetching, batched locate, 10-mer precomputation, ungapped fast paths, and reduced mate rescue each contribute to speed/accuracy; provide controlled ablations.
- Potential integration with learned indexes is unexplored
- BWA-MEME shows learned-index gains for seeding; assess whether learned structures can accelerate SMEM discovery in this framework without sacrificing accuracy.
- GPU/accelerator support not considered
- Given DP bottlenecks and successful GPU ports of related mappers, explore GPU/FPGA offload and hybrid CPU–GPU scheduling for further speedups.
- Real-data robustness across environments and implementations
- Rust rewrite shows ±20% variability on long reads across environments; provide reproducibility analyses (compiler flags, CPUs, OS) and a comprehensive test suite beyond “several datasets.”
- Repetitive-region MAPQ downweighting and user controls
- If reduced effort in repeats is retained, expose tunable controls and document how MAPQ is adjusted in repeats to prevent overconfident mappings.
- Chain-scoring behavior with variable-length seeds not deeply evaluated
- Assess mis-joins and chaining errors in SV-rich regions and long reads; compare to minimap2’s heuristics under different seed distributions and read error profiles.
- Library and insert-size assumptions in mate rescue
- The q-mer prefilter and reduced rescue candidates may hurt sensitivity for atypical libraries (broad insert distributions, contamination); quantify robustness and consider adaptive thresholds.
Practical Applications
Overview
Minibwa is a fast, FM-index–based genomic read aligner that merges BWA-MEM’s variable-length seeding with minimap2’s chaining and SIMD-accelerated base alignment. Key innovations include a batched/prefetched SMEM-finding algorithm and locate operation, more frequent ungapped fast paths, reduced effort in highly repetitive regions, adaptive parameterization across read lengths, and native support for directional bisulfite sequencing (BS-seq). It delivers roughly 4× speed over BWA-MEM and >2× over BWA-MEM2 at comparable accuracy, supports both short and accurate long reads (HiFi/ONT), and outperforms common BS-seq mappers in speed while maintaining high accuracy.
Below are practical applications grouped by time horizon, with sectors, potential tools/workflows, and assumptions/dependencies.
Immediate Applications
- Faster drop-in alignment for clinical and research WGS variant calling
- Sectors: Healthcare (clinical genomics), biotech/CROs, academia.
- Tools/workflows: Substitution for BWA-MEM/BWA-MEM2 in GATK Best Practices, DeepVariant, Strelka2, DRAGEN-like CPU pipelines; integration into WDL/CWL/Nextflow (e.g., nf-core/sarek).
- Impact: 2–4× faster mapping at comparable small-variant accuracy; reduced cloud/on-prem compute costs and turnaround time.
- Assumptions/dependencies: Validation for clinical use; MAPQ and alignment heuristics not bit-identical to BWA-MEM; current lack of alternate-contig support; typical peak RAM <20 GB; SSE4.1 (x86) or NEON (ARM) available.
- Unified aligner for mixed read lengths (short reads, SBX mid-length, accurate long reads)
- Sectors: Core sequencing facilities, biotech, academia.
- Tools/workflows: Single aligner choice for Illumina + Roche SBX + HiFi/ONT runs; simplified pipeline maintenance; adaptive parameterization by read length.
- Impact: Eliminates aligner switching; reduces operational complexity and errors.
- Assumptions/dependencies: Mixed datasets routed through one pipeline; long-read performance competitive with minimap2 confirmed by benchmarking.
- Accelerated BS-seq (WGBS/targeted) alignment for methylation profiling
- Sectors: Epigenomics (research and translational), biotech, oncology.
- Tools/workflows: Replacement for Bismark/BISCUIT/BWA-Meth in pipelines with MethylDackel, Methyldackel+segmentation tools, or bespoke methylation callers.
- Impact: Several-fold speedups over BISCUIT/BWA-Meth and >10× over Bismark; improved pairing logic and asymmetric scoring reduces miscalls.
- Assumptions/dependencies: Directional BS-seq supported natively; non-directional BS-seq may require settings or future support; larger index (four converted genomes) but peak RAM remains <20 GB.
- Faster Hi-C read alignment
- Sectors: 3D genomics (academia/biotech).
- Tools/workflows: Swapping BWA-MEM for minibwa in Juicer, HiC-Pro, or in-house pipelines.
- Impact: Reduced compute time for large Hi-C datasets; improved throughput.
- Assumptions/dependencies: Mate rescue typically disabled in Hi-C; parameter tuning may be needed per kit.
- Cost and energy reduction for population-scale sequencing projects
- Sectors: Public health, population genomics (e.g., All of Us, UK Biobank), national genomics initiatives.
- Tools/workflows: Backend alignment in batch and streaming settings using Cromwell/Terra, DNAnexus, Seven Bridges, Nextflow Tower.
- Impact: Large aggregate cost savings; lower energy footprint and carbon emissions.
- Assumptions/dependencies: Sufficient parallelism; I/O infrastructure; consistent MAPQ handling downstream.
- ARM-first cloud deployments (cost/performance gains)
- Sectors: Cloud bioinformatics platforms, DevOps.
- Tools/workflows: Running minibwa on AWS Graviton/OCI Ampere instances; containerized deployment (Docker/Singularity) with NEON support.
- Impact: Additional cost benefits beyond algorithmic speedup.
- Assumptions/dependencies: Build and test on ARM; performance verified on selected instance types.
- Rapid QC and triage of sequencing runs
- Sectors: Sequencing cores, LIMS operations.
- Tools/workflows: Early-stage alignment for insert-size, coverage, contamination checks; real-time run assessment.
- Impact: Faster go/no-go decisions; reduced wasted sequencing and turnaround delays.
- Assumptions/dependencies: Lightweight QC pipelines; sufficient RAM on QC nodes.
- Long-read upstream alignment for SV and assembly pipelines
- Sectors: Structural variant analysis, de novo assembly (research/biotech).
- Tools/workflows: Use minibwa instead of minimap2 for alignment prior to SV callers (Sniffles, pbsv, SVIM) or assembly polishing.
- Impact: Comparable accuracy with modest speed gains; simplified toolchain if unifying aligners across data types.
- Assumptions/dependencies: SV calling parity depends on downstream tools’ sensitivity to subtle alignment differences; validation recommended.
- High-throughput reprocessing of legacy archives
- Sectors: Data repositories (ENA/SRA, institutional archives), biobanks.
- Tools/workflows: Re-aligning historical data to new references (e.g., T2T-CHM13, updated GRCh), rebaselining variant sets.
- Impact: Lower reprocessing costs; faster turnaround for harmonization projects.
- Assumptions/dependencies: Compute/storage budget; standardized output requirements.
- Method transfer to other FM-index–based tools
- Sectors: Bioinformatics software engineering.
- Tools/workflows: Adoption of batched locate and batched SMEM with prefetch in other aligners/indexed tools (e.g., future Bowtie/BWA variants, FM-index search libraries).
- Impact: Broad speedups across the FM-index ecosystem.
- Assumptions/dependencies: Availability of source integration; careful correctness/latency tuning.
Long-Term Applications
- Ecosystem-wide replacement of BWA-MEM in best-practice pipelines
- Sectors: Healthcare, biotech, academia, public genomics.
- Tools/workflows: Official GATK/nf-core recommendations updated; packaged modules in Bioconda/Conda-forge; cloud marketplace AMIs.
- Impact: Standardization; sustained cost reductions and maintainability.
- Assumptions/dependencies: Community consensus; completion of redesigned alternate-contig support; broad validation across cohorts.
- Real-time/on-instrument alignment and near–real-time analysis
- Sectors: Rapid diagnostics (NICU/ICU), outbreak response, field genomics.
- Tools/workflows: Streaming pipelines coupling basecalling and minibwa; immediate variant calling on partial data.
- Impact: Hours shaved off end-to-end turnaround.
- Assumptions/dependencies: Tight I/O integration with sequencers; robust streaming MAPQ calibration; hardware constraints on embedded CPUs.
- GPU/FPGA or heterogeneous compute acceleration of batched FM-index operations
- Sectors: High-performance computing, cloud providers.
- Tools/workflows: Offloading batched locate/SMEM search and DP kernels; hybrid CPU+accelerator designs.
- Impact: Additional throughput and cost savings for petabyte-scale operations.
- Assumptions/dependencies: Algorithmic refactoring for accelerators; ROI depends on dataset scale and hardware availability.
- Pangenome/graph-aware extensions
- Sectors: Advanced genomics (reference diversity initiatives).
- Tools/workflows: Integrating variable-length seeding and minimap2-style chaining into graph indices (e.g., GCSA2/GBWT successors).
- Impact: Improved mapping accuracy across diverse ancestries and structurally variant loci.
- Assumptions/dependencies: New indexing schemes; memory and engineering complexity; community adoption of graph references.
- Centromere- and repeat-aware mapping policies with T2T references
- Sectors: Research and clinical genomics.
- Tools/workflows: Heuristics that exploit T2T-CHM13 and future assemblies to reduce false high-MAPQ in structurally divergent repeats; configurable compute skiplists.
- Impact: More reliable confidence estimates; better downstream interpretation.
- Assumptions/dependencies: Availability of high-quality per-sample SV-aware references; calibration studies.
- Scalable single-cell whole-genome and single-cell methylome alignment
- Sectors: Single-cell genomics (research/biotech).
- Tools/workflows: High-throughput alignment modules for scWGS/scWGBS; cost control in massive cell atlases.
- Impact: Feasible processing at new scales; budget containment.
- Assumptions/dependencies: Barcoding-aware pre/post-processing; memory and I/O optimized batching.
- Integrated methylation-aware variant calling and epigenetic assays
- Sectors: Oncology diagnostics, liquid biopsy, translational research.
- Tools/workflows: Pipelines that jointly model sequencing and methylation errors leveraging minibwa’s asymmetric scoring for BS-seq.
- Impact: Improved calls in methylation-rich contexts; consolidated workflows.
- Assumptions/dependencies: Development of joint callers; clinical validation.
- Policy and procurement guidance for greener, lower-cost genomics
- Sectors: Funding agencies, national labs, public health.
- Tools/workflows: Best-practice documents encouraging high-efficiency mappers; carbon accounting in grant reporting.
- Impact: Reduced operational costs and emissions at scale.
- Assumptions/dependencies: Auditable benchmarks; alignment accuracy consensus; governance updates.
Cross-cutting assumptions and dependencies
- Reference/index requirements: FM-index build (BWT ~3 GB for human) plus working memory; larger for BS-seq converted references.
- Hardware: SIMD support (SSE4.1 on x86, NEON on ARM); benefits increase with memory bandwidth and cache performance; prefetch-friendly CPUs.
- Software compatibility: SAM/BAM tags and MAPQ distributions may differ slightly from BWA-MEM; downstream tools should tolerate these differences or be recalibrated.
- Data characteristics: Performance/accuracy trade-offs in highly repetitive regions are intentional; for studies focused on such regions, parameter tuning or alternate strategies may be needed.
- Regulatory/validation: Clinical adoption requires method validation, documentation of equivalence (or superiority), and change-control processes.
These applications leverage minibwa’s speed, cross–read-length versatility, and BS-seq support to reduce costs and turnaround times, enable new scales of analysis, and catalyze broader algorithmic improvements across the read mapping ecosystem.
Glossary
- 3-base strategy: A bisulfite-seq mapping approach that reduces the alphabet by treating C↔T (and G↔A) conversions specially so aligners can handle methylation-induced changes. "uses the so-called 3-base strategy to align directional BS-seq data."
- Acrocentric short arms: The short arms of acrocentric human chromosomes, rich in repeats and challenging for accurate read placement. "centromeres or acrocentric short arms."
- Affine gap penalty: An alignment scoring scheme with a gap-open cost plus a smaller per-base gap extension cost. "BWA-MEM uses the standard affine gap penalty"
- Alternate contigs: Supplementary reference sequences representing alternate haplotypes/structures for difficult regions. "the support of alternate contigs in the reference genome"
- Asymmetric scoring matrix: An alignment scoring scheme where certain mismatches (e.g., C→T in bisulfite data) are penalized differently from their reverse. "an asymmetric scoring matrix that permits C-to-T mismatches and penalizes T-to-C mismatches."
- AVX2: An x86 SIMD instruction set extension enabling wider vector operations for performance. "We experimented an AVX2-based implementation but did not see clear improvement."
- Backward extension: Extending a pattern to the left in FM-index search to refine its interval. "tests if P[i,i+ℓ) is a match with backward extension"
- Backward search: The classical FM-index procedure that scans a pattern from right to left to find its occurrences. "Take the standard backward search as a case study."
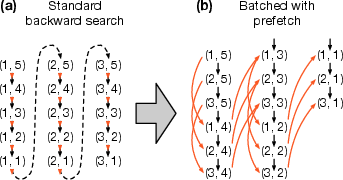
- Batched locate operation: A latency-hiding method that resolves many suffix array positions to text coordinates in batches. "Batched locate operation"
- Bisulfite sequencing (BS-seq): A technique to profile DNA methylation by converting unmethylated cytosines to thymines before sequencing. "bisulfite sequencing (BS-seq) reads."
- Burrows-Wheeler Transform (BWT): A reversible text transform underpinning FM-indexes that groups similar characters to enable compression and fast queries. "B is the Burrows-Wheeler Transform (BWT) of T."
- Centromere: Highly repetitive chromosomal regions essential for segregation, often hard to align reads uniquely. "Exhaustive alignment in centromeres is a waste of effort"
- Chaining: Linking seeds into co-linear chains across read and reference to propose candidate alignments. "the minimap2 chaining algorithm"
- DeepVariant: A deep-learning-based small-variant caller from aligned reads. "DeepVariant v1.10.0"
- Directional bisulfite sequencing: BS-seq libraries that preserve strand orientation, constraining valid conversion patterns. "directional BS-seq data"
- Double-strand suffix array interval (ds-interval): A tuple representing the FM-index interval for a pattern across both strands of a bidirectional index. "The double-strand suffix array interval (ds-interval) of P"
- Dual-gap penalty: A DP scoring model with two gap regimes (e.g., for short vs long gaps), each with its own costs. "with dual-gap penalty"
- Dynamic programming (DP): The standard matrix-filling method for sequence alignment (e.g., Smith–Waterman/Needleman–Wunsch). "skip full dynamic programming"
- FM-index: A compressed full-text index based on the BWT that supports fast substring queries with low memory. "To reduce memory with FM-index, we only store S(i) when i is a multiple of parameter r."
- Forward extension: Extending a pattern to the right in FM-index search to refine or complete an SMEM. "we use forward extension to find the end of the SMEM"
- GIAB Q100: A Genome in a Bottle high-confidence benchmark set filtered to Q100 regions for variant evaluation. "the GIAB Q100 ground truth v1.1"
- Hi-C: A sequencing assay capturing chromatin contacts; here, paired-end reads with atypical insert distributions. "Hi-C reads"
- HiFi reads: High-fidelity long reads (e.g., PacBio) with low error rates used for accurate long-read alignment. "HiFi reads simulated from the diploid HG002 genome"
- Latency hiding: Overlapping memory accesses and computation (often via batching) to reduce stalls from cache misses. "Latency hiding is an effective technique."
- Learned indices: ML-based models that approximate index structures (e.g., CDFs) to speed up lookups. "BWA-MEME further speeds up BWA-MEM2 seeding with learned indices."
- LF-mapping: The last-to-first mapping in BWT-based indexes that advances positions during backward/forward extension. "is the LF-mapping."
- Locate operation: The step that resolves FM-index interval positions to actual text (chromosomal) coordinates using sampled SAs. "The “locate” operation finds the chromosomal positions of [k,k+s) given a sampled suffix array."
- Mapping quality: A probabilistic score (often Phred-scaled) estimating the confidence that a read is correctly placed. "stratified by mapping quality."
- Mate rescue: Attempting to align an unmapped mate near the mapped read using local alignment. "skip unnecessary mate rescue"
- N50: A length statistic where half of total bases are in reads at least this long. "the N50 read length of this duplex SBX dataset is 243 bp"
- NEON intrinsics: ARM SIMD intrinsics used to vectorize compute kernels. "NEON intrinsics"
- Prefetching: Explicitly requesting memory into cache ahead of use to reduce latency. "We can prefetch the data at this address"
- q-mer: A substring of length q used for fast filtering or seeding in alignment pipelines. "prefilters the read-reference pair by testing q-mer matches (q=7)."
- ropebwt3: An algorithm/tooling for BWT/SMEM operations optimized for speed and batching. "adapts the ropebwt3 algorithm for finding supermaximal exact matches (SMEMs)."
- SBX: Roche’s emerging sequencing technology producing reads between short and long regimes. "Roche's SBX sequencing technology"
- Sentinel: A special end-of-text symbol added to reference strings in indexing. "a trailing sentinel $\$$"
- SIMD: Single Instruction Multiple Data vectorization to operate on multiple data elements per instruction. "Single Instruction Multiple Data (SIMD) instructions."
- Smith–Waterman alignment: The classic local alignment algorithm used for sensitive pairwise alignment. "Smith-Waterman alignment"
- SSE4.1 intrinsics: x86 SIMD intrinsic functions targeting the SSE4.1 instruction set for vectorized compute. "SSE4.1 intrinsics"
- Suffix array (SA): An array of starting positions of lexicographically sorted suffixes of a text. "The suffix array (SA) of T is an integer array S"
- Supermaximal exact match (SMEM): An exact match on the query that is not contained in any longer exact match on the query. "supermaximal exact matches (SMEMs)."
- Ungapped alignment: Alignment allowing mismatches but no insertions/deletions; a fast heuristic before DP. "Minibwa first tries ungapped alignment."
- Watson–Crick complement: The DNA base-complement rule (A↔T, C↔G), used for reverse-complement handling. "is the Watson-Crick complement of a."
- Whole-Genome Sequencing (WGS): Sequencing that targets the entire genome, producing large volumes of reads. "Whole-Genome Sequencing (WGS)"
Collections
Sign up for free to add this paper to one or more collections.

