- The paper introduces a novel neighborhood-constrained mixup method that improves spatial gene expression imputation by aligning local transcriptomic features.
- It leverages KNN-based spatial neighborhoods and Gaussian kernel weighting to ensure biologically plausible augmentation that preserves tissue architecture.
- Experiments demonstrate significant reductions in MSE and gains in Pearson correlation across diverse tissue datasets, validating the method's effectiveness.
SNR-ST-Mix: Sample-specific Neighborhood Regression Mixup for Augmented Spatial Transcriptomics Imputation
Introduction and Motivation
Spatial transcriptomics (ST) enables high-throughput measurement of gene expression within tissue architecture, facilitating molecular insights in complex biological systems. Despite its transformative impact, ST data generation suffers from high financial and operational costs, technical noise, low spatial resolution, and sparse spot coverage, which collectively hinder robust modeling of fine anatomical structures and generalizable transcriptomic patterns. Deep neural networks have demonstrated efficacy in spatial gene expression imputation from histology, yet their sample-level training is fundamentally bottlenecked by dataset size and lack of biology-aware augmentation. Previous data augmentation techniques—such as mixup, CutMix, and related regularization strategies—are fundamentally designed for classification and ignore critical spatial and molecular dependencies in ST regression, leading to suboptimal learning and biologically implausible interpolations.
Methodological Framework: SNR-ST-Mix
SNR-ST-Mix introduces a regression-oriented, spatially and transcriptionally informed augmentation regime tailored for ST imputation. Unlike traditional mixup that interpolates indiscriminately across all samples, SNR-ST-Mix generates synthetic examples by constraining mixup to a spot’s k-nearest spatial neighbors and adaptively weighting partners according to transcriptomic similarity. The patch pairs generated in this way are substantially more likely to reflect biologically plausible transitions, enforce spatial smoothness, and preserve underlying anatomical microstructure.
The high-level pipeline can be summarized as follows:
- Patch Extraction: The WSI is divided into patches mapped to spatial spots, with expression vectors available for assayed locations.
- Spatial Neighborhood Construction: For each spot, a KNN search identifies its K closest spatial neighbors, guaranteeing local context.
- Expression Similarity-based Partner Selection: A Gaussian kernel on L2 distance in gene expression space modulates mixup pairing probabilities, biasing toward transcriptomically coherent partners.
- Neighbor-Constrained Mixup: Convex combinations of both image patches and gene expression vectors are constructed (weighted by Beta-distributed λ) to expand sample diversity while maintaining biological and spatial consistency.
- Learning Objective: The loss function couples MSE with two structure-aware regularizers (mixup consistency, edge alignment) and a mild correlation prior for robust regression.
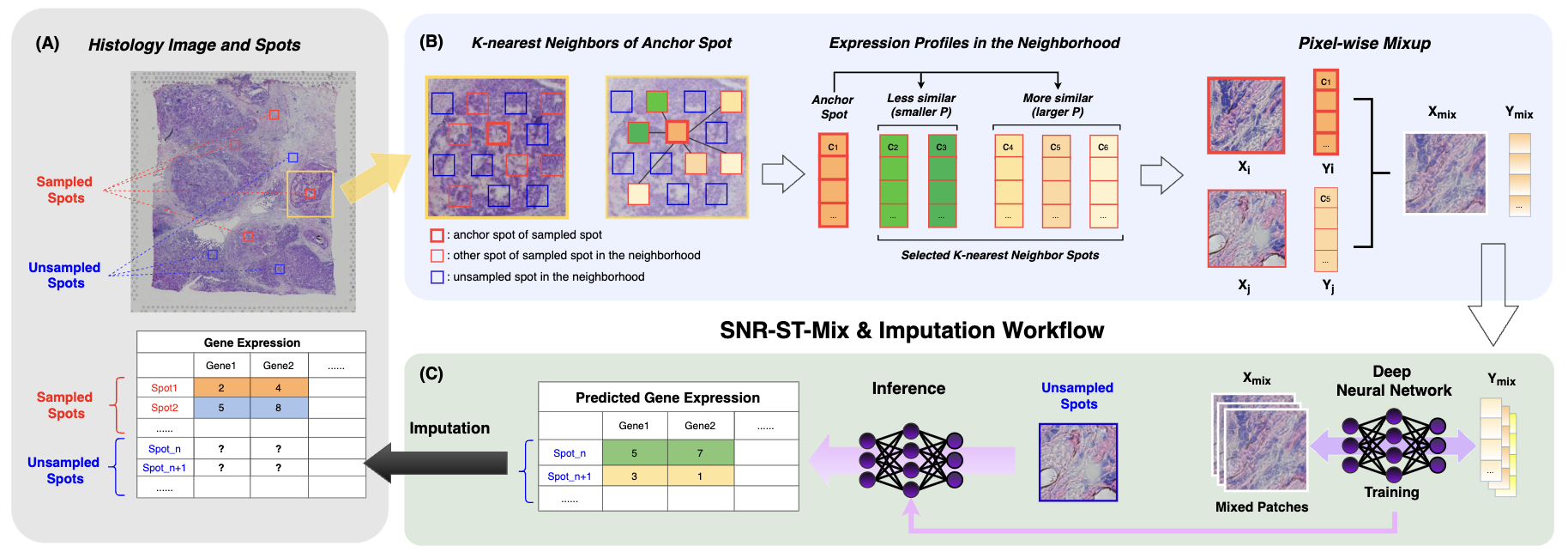
Figure 1: Overview of SNR-ST-Mix, demonstrating patch extraction, KNN-based spatial neighborhood, similarity-weighted mixup pairing, and downstream regression for ST imputation.
Empirical Results
Qualitative Analysis
Visualization experiments reveal that SNR-ST-Mix provides notable denoising, enhanced preservation of spatial boundaries, and improved recovery of region-specific expression gradients compared to vanilla mixup, CutMix, and basic augmentations. Sharper boundaries and improved morphologically guided prediction are particularly evident in tumor datasets with pronounced regional heterogeneity.
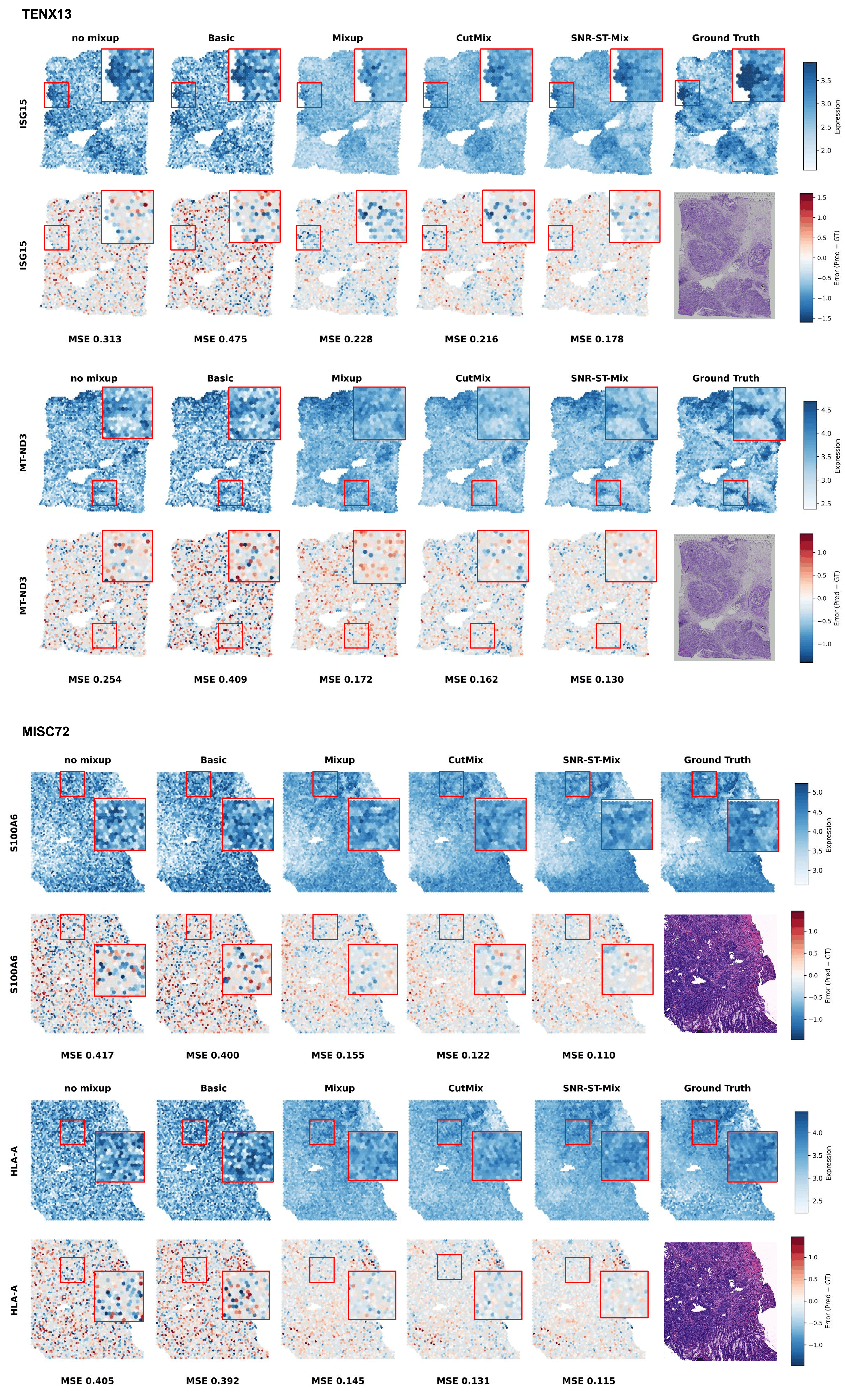
Figure 2: Comparisons of predicted gene expression maps and associated error heatmaps across methods. SNR-ST-Mix yields the most accurate and spatially coherent predictions.
Quantitative Analysis
SNR-ST-Mix achieves consistently superior performance across eight ST datasets (spanning breast, bowel, ovary, prostate cancer, and healthy heart), as measured by MSE, MAE, and Pearson correlation coefficients. For example, in TENX13 (breast cancer), the MSE drops from 0.2074 (baseline) to 0.1886, with a corresponding PCC increase from 0.4180 to 0.4748. Improvements are similarly significant in other tissue types and for nearly all genes.
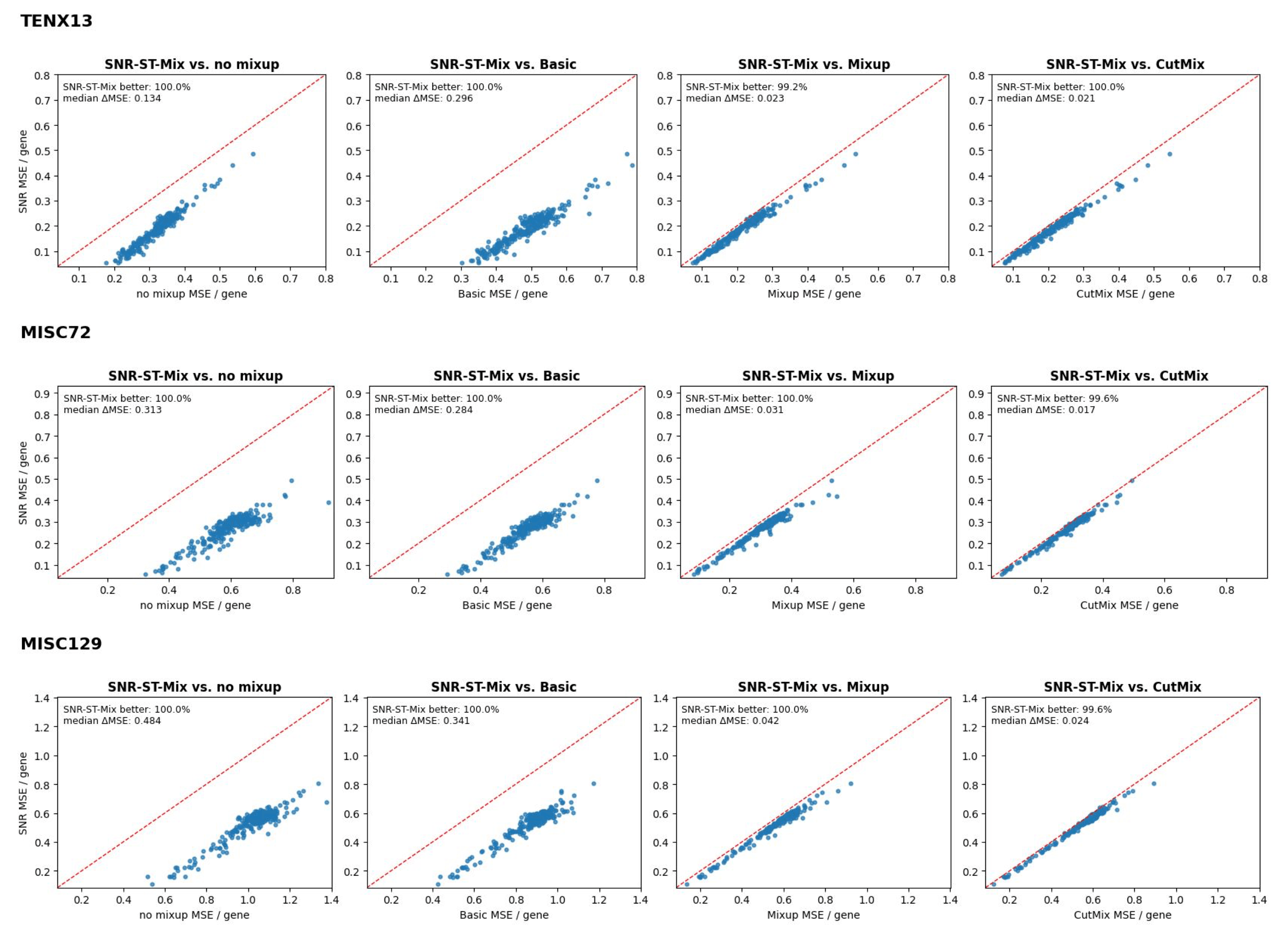
Figure 3: Scatter plots comparing per-gene MSE values under SNR-ST-Mix vs. baseline augmentations; SNR-ST-Mix outperforms for nearly all genes across datasets.
Ablation Studies and Hyperparameter Robustness
Progressively integrating KNN, label similarity, and loss regularization components into vanilla mixup produces additive gains. Joint spatial and transcriptomic conditioning substantially outperform either constraint alone. Further, SNR-ST-Mix demonstrates robustness to a broad range of values for the number of neighbors (K) and kernel bandwidth (σ), indicating practicality for heterogeneous datasets without the need for exhaustive tuning.
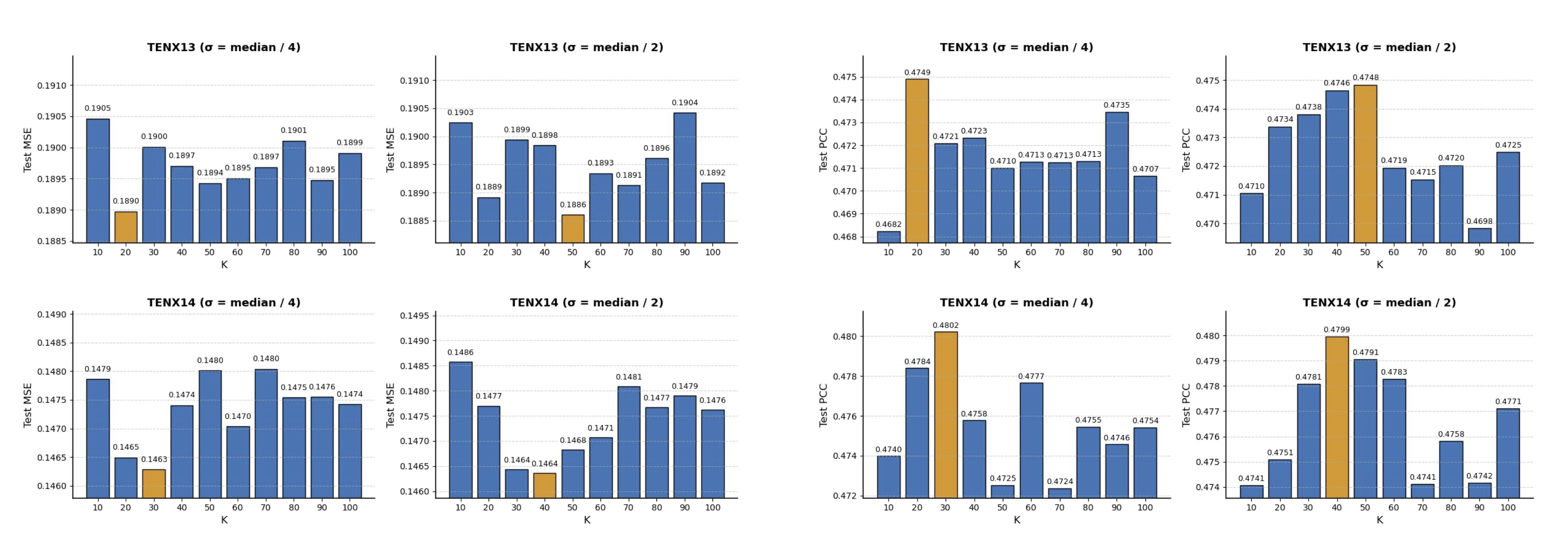
Figure 4: Ablation on K and σ hyperparameters shows resilience of SNR-ST-Mix’s performance to parameter variations (test MSE and PCC plotted).
Theoretical Implications
SNR-ST-Mix’s design is informed by a Lipschitz prior on the latent spatial gene expression field. By limiting mixup to local spatial neighborhoods and ensuring transcriptomic alignment, the method preserves smoothness priors and mitigates variance induced by spot-specific, heteroscedastic measurement noise. The explicit bias toward local linearity (via the consistency regularizer) and graph Laplacian smoothness (via edge alignment) yields a more stable predictive manifold, better generalization, and improved biological fidelity.
Practical Impacts and Future Directions
SNR-ST-Mix’s architecture-agnostic, computationally lightweight implementation makes it ideal for integration into diverse histology-guided ST pipelines and for scaling to single-sample or multi-sample imputation scenarios where data diversity and labeling are limiting factors. Notably, the method's advantages are most pronounced in tissues with high local heterogeneity (e.g., tumor microenvironments), but it remains competitive across structurally homogeneous contexts. The augmentation paradigm introduced here generalizes in principle to region-level mixing, CutMix variants, and other multimodal settings, suggesting possible adaptation for classification, segmentation, and downstream tissue phenotyping.
Potential future extensions include:
- Adaptive or learnable neighborhood scales/enhanced local heterogeneity modeling
- Augmentation-aware integration with deconvolution algorithms for single-cell or subcellular ST platforms (e.g., MERFISH, Xenium)
- Broader application beyond gene expression imputation, including clinically relevant outcome prediction and cross-modality translation tasks
Conclusion
SNR-ST-Mix establishes a principled, regression-compatible data augmentation framework for spatial transcriptomics, explicitly aligning mixup sampling with biological and tissue-structural constraints. The approach rigorously overcomes the pitfalls of classification-inspired augmentations in continuous ST regression, ensuring synthetic examples are plausible, smooth, and robust. Experimental evidence demonstrates that SNR-ST-Mix systematically outperforms existing augmentations in diverse ST settings, offering a scalable solution to sample-specific model limitations. As ST continues its advance toward higher resolution and multimodal measurements, SNR-ST-Mix sets the foundation for next-generation, biology-aware augmentation frameworks with broad applicability across computational pathology and spatial omics.