- The paper introduces an all-electron dynamical BSE framework that incorporates frequency-dependent screening using numerical atom-centered orbitals.
- It employs an effective dielectric function method and symmetry-adapted IBZ mapping to reduce computational overhead while preserving local-field effects.
- Validation on crystalline naphthalene demonstrates sub-30 meV accuracy in exciton binding energies and a noticeable redshift in excitation energies.
All-electron Dynamical Bethe-Salpeter Equation for Extended Systems with Atom-centered Orbital Basis Set
Overview and Motivation
The Bethe-Salpeter Equation (BSE) is a cornerstone for ab initio calculations of neutral excitations, capturing excitonic effects in the framework of many-body Green’s function theory. Traditionally, static approximations are used for the screened Coulomb interaction kernel, neglecting its frequency dependence. However, for systems with large exciton binding energies—including organic crystals, low-dimensional semiconductors, and small molecular solids—dynamical screening effects become non-negligible, rendering static BSE treatments inadequate.
This paper introduces a rigorous all-electron dynamical BSE framework employing numerical atom-centered orbitals (NAOs) for extended systems. The authors generalize the effective dielectric function approach originally devised for plane-wave bases to an NAO-based formalism, thus enabling dynamical BSE calculations on extended systems in a computationally efficient and systematically improvable all-electron setting (2606.08350).
The foundational challenge addressed is the inclusion of frequency-dependent screened Coulomb interactions in BSE, which formally leads to a coupled non-linear eigenvalue problem. The static approximation—commonly justified for small exciton binding energies relative to the plasmon frequency—breaks down for strongly bound excitons.
The authors adopt the effective dielectric function method, building upon the Shindo approximation, which decouples frequency dependencies and circumvents direct frequency integration. The interaction kernel in the dynamical BSE is replaced by an effective, frequency-independent dielectric function parameterized by the exciton binding energy, allowing practical computation with significant accuracy gains over the static approach.
Moreover, the theoretical exposition details the translation of this method to the NAO basis: the Coulomb and dielectric operators are expanded using auxiliary basis functions (ABFs), and all necessary matrix elements are constructed via resolution-of-identity techniques. Spectral decomposition and nonlinear functional transformation of the dielectric matrix in the ABF basis rigorously treat local-field effects, which are neglected in plane-wave diagonal approximations.
Symmetry Adaptation and Brillouin Zone Mapping
A significant computational bottleneck in BSE calculations is the dense Brillouin zone (BZ) sampling required for convergence, especially for GW-based quasiparticle energies. To mitigate this, the paper implements a symmetry-adapted mapping from the irreducible Brillouin zone (IBZ) to the full BZ, drastically reducing redundant evaluations of the screened interaction matrices. The symmetry transformation matrices efficiently propagate dielectric and Coulomb tensors for both static and dynamical BSE, yielding substantial reductions in computational effort without loss of accuracy.
Validation and Numerical Demonstration
Crystalline naphthalene, a monoclinic organic crystal with pronounced excitonic effects, serves as the primary benchmark.
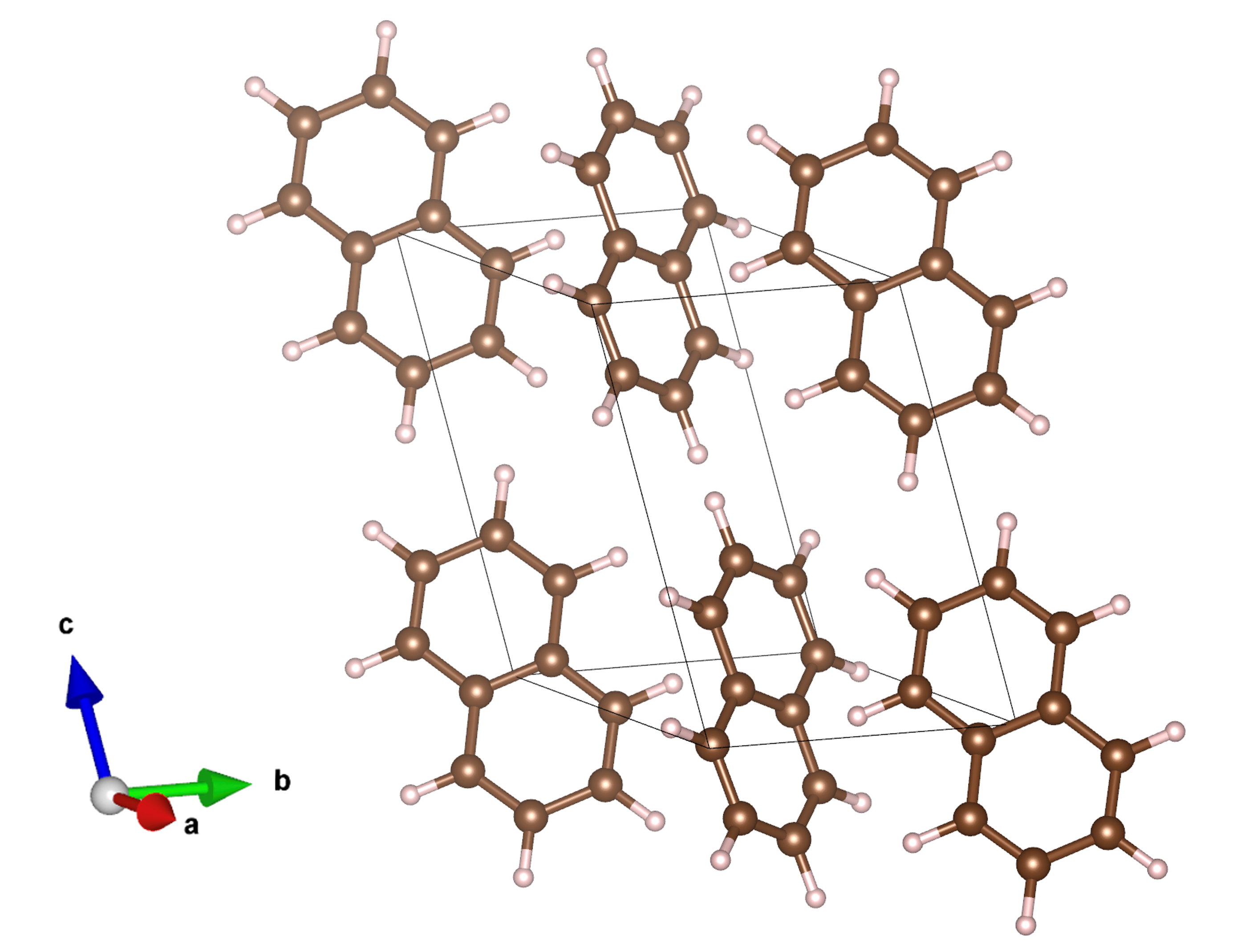
Figure 1: Crystal structure of monoclinic naphthalene (C10H8) illustrating its herringbone packing.
The NAO-based dynamical BSE implementation is validated against prior plane-wave results for DFT+Δ calculations, demonstrating sub-30 meV agreement in exciton binding energies and optical gaps. The dynamical BSE introduces a ∼0.1 eV redshift in excitation energies for naphthalene, consistent with previous perturbative and exact-diagonalization studies.
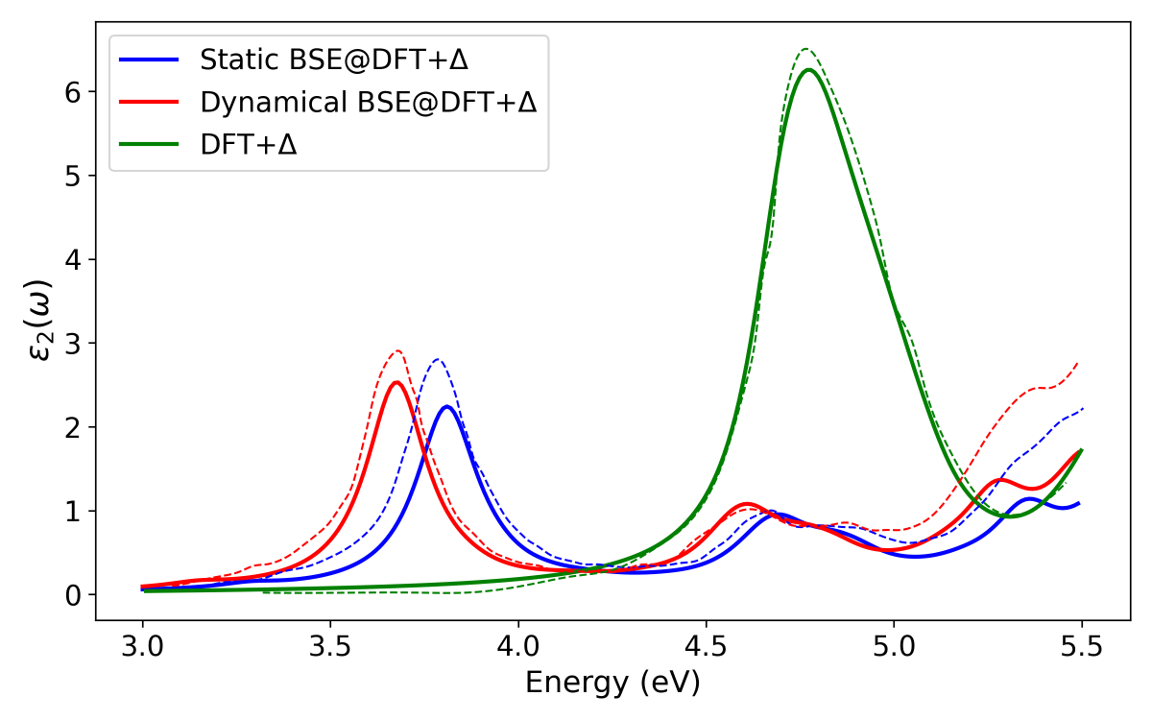
Figure 2: Comparison of optical absorption spectra in the b direction: DFT+Δ, static BSE@DFT+Δ, and dynamical BSE@DFT+Δ using an exciton binding energy of 1.06eV.
The symmetry-adapted IBZ mapping yields identical optical spectra compared to full BZ calculations but with a ∼70% reduction in sampled points for naphthalene's C10H80 grid.
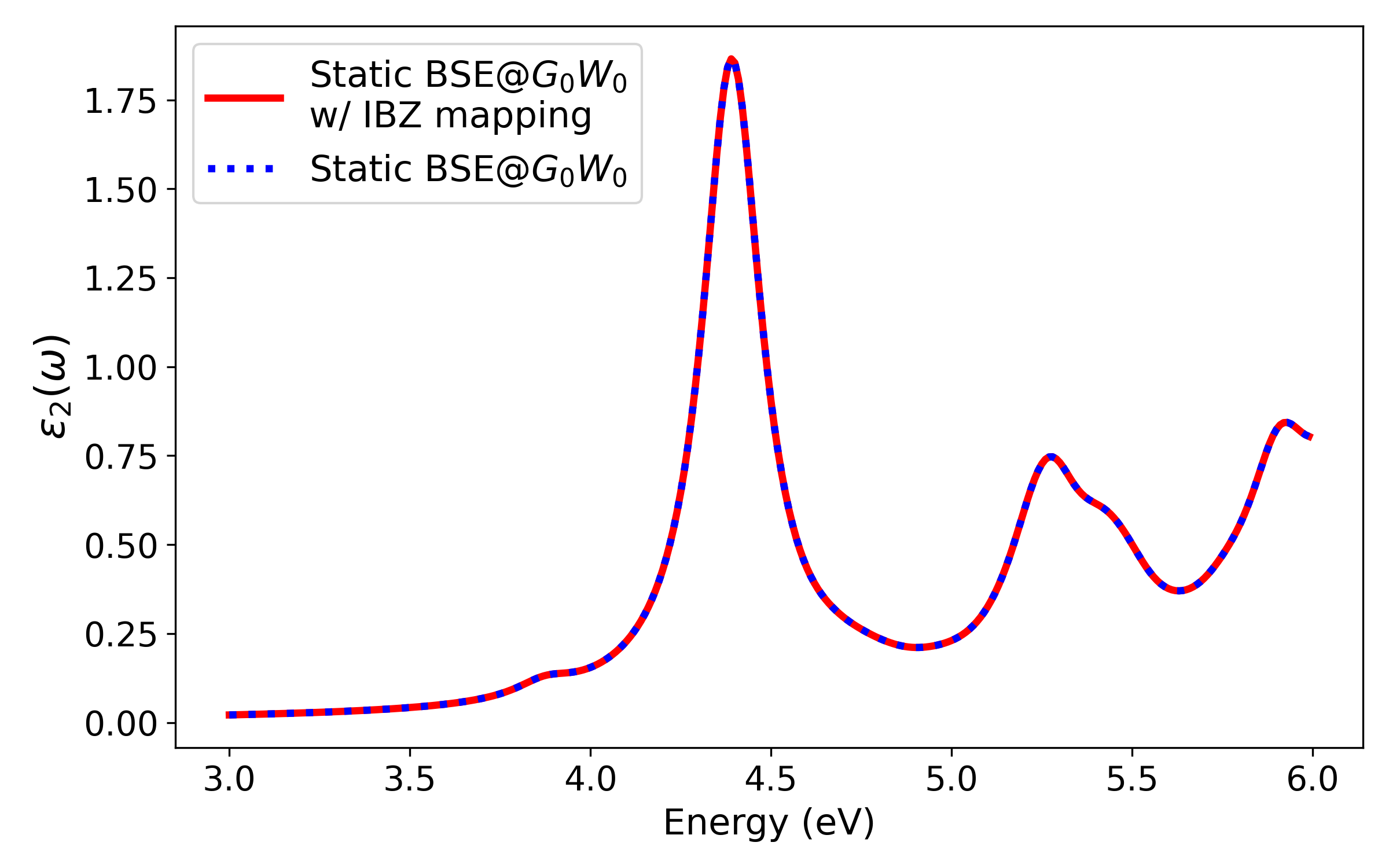
Figure 3: Optical absorption spectra comparison for static BSE@C10H81, illustrating identical results with and without the IBZ mapping scheme.
The authors further demonstrate the method in combination with C10H82 quasiparticle energies. Static and dynamical BSE@C10H83 spectra show that dynamical screening increases the exciton binding energy by C10H84 eV and yields a redshift in optical excitation peaks, aligning with analytical expectations for systems with large exciton binding.
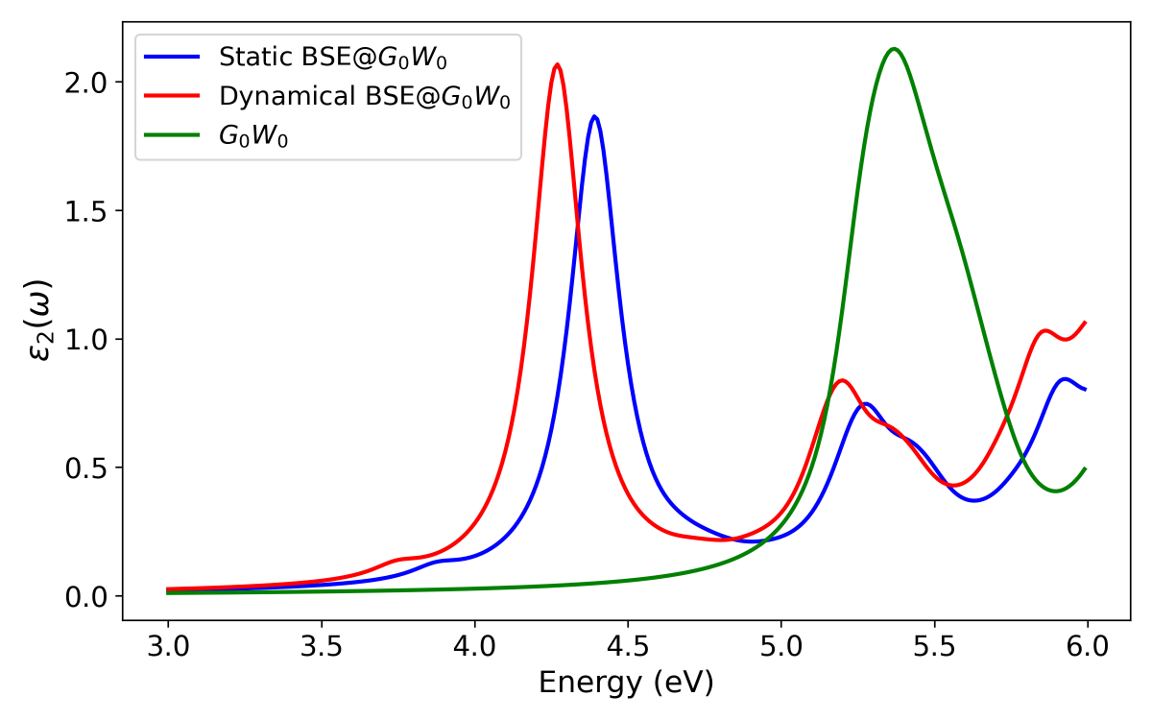
Figure 4: Optical absorption spectra for C10H85, static BSE@C10H86, and dynamical BSE@C10H87. The dynamical BSE introduces a notable redshift.
Implications and Outlook
The demonstrated NAO-based dynamical BSE framework achieves systematic all-electron accuracy and efficiently incorporates dynamical screening effects critical for strongly bound excitonic systems. The symmetry-adapted IBZ mapping offers a scalable path for high-throughput BSE calculations on extended periodic materials, drastically reducing computational overhead.
The capability for full-matrix treatment of dielectric screening (including non-diagonal local-field effects) positions this approach for future expansion: energy-specific iterative eigensolvers, C10H88-grid interpolation, and Wannier-based techniques can be layered onto this foundation. The methodology enables rigorous study of optical properties, excitonic phenomena, and core excitations in complex organic and inorganic solids, facilitating quantitative spectroscopy, predictive design of optoelectronic systems, and systematic benchmarking of many-body perturbation theory.
Conclusion
This work presents a robust all-electron, NAO-based dynamical BSE method for extended systems, generalizing the effective dielectric function approach and systematically validating its accuracy for a paradigmatic organic crystal. The integration of symmetry-adapted zone mapping, full local-field effects, and efficient auxiliary basis expansion constitutes a significant technical advance in ab initio excited-state electronic structure. Future developments along the lines of iterative solvers, further symmetry exploitation, and advanced interpolation promise to expand the scope and scalability of BSE@C10H89 calculations for increasingly complex material systems.

