- The paper introduces AF_Cache, which integrates GPU-accelerated batched MSA generation, aggressive monomer feature caching, and bucketing to eliminate redundant computations in PPI prediction.
- It achieves significant speedups, including a 13-fold reduction in MSA runtime and an average 90-second decrease per protein pair, optimizing resource use in AlphaFold pipelines.
- The scalable workflow supports both AF2 and AF3, maintaining high structural accuracy with strong ipTM score correlations for experimentally validated protein interactions.
AF_Cache: Accelerating High-Throughput AlphaFold-Based Protein–Protein Interaction Prediction
Introduction and Motivation
The accurate elucidation of protein–protein interactions (PPIs) is essential for decoding cellular mechanisms and biomolecular network architectures. The advent of neural network-driven methods, particularly AlphaFold2 (AF2) and AlphaFold3 (AF3), has greatly increased the precision of in silico complex structure predictions. However, their default pipelines remain computationally prohibitive for proteome-scale or network-wide applications, largely due to the repeated and CPU-bound generation of MSAs, and, in the case of AF2, expensive JAX model recompilations for each target of varying sequence length.
AF_Cache presents a systematic pipeline engineered to address these computational bottlenecks by integrating GPU-accelerated batched MSA generation, aggressive caching of monomeric features, and efficient bucketing and padding schemes to minimize JAX recompilation events. The pipeline is realized as a robust, reproducible workflow via Nextflow, supporting both AF2 and AF3, and is specifically designed for scalable, high-throughput PPI prediction settings.
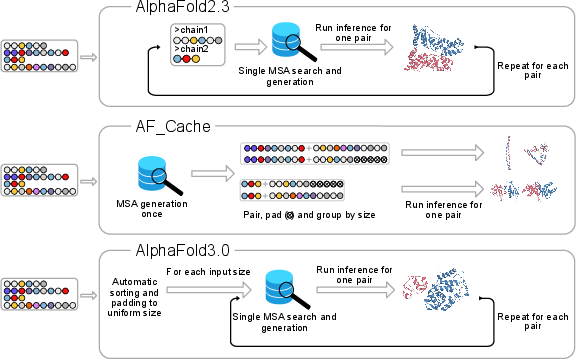
Figure 1: Overview of AF_Cache and AlphaFold pipelines highlighting batched MSA generation, feature caching, and size bucketing to optimize throughput.
Pipeline Design and Computational Optimizations
Batched, GPU-Accelerated MSA Generation
MSA generation is accelerated by employing MMseqs2-GPU, enabling global batched searches across the target set. Both UniRef and environmental database search stages are parallelized, with CPU and GPU operations overlapping, exploiting otherwise idle hardware periods. AF_Cache circumvents per-pair MSA recomputation in multimers: monomeric MSAs and template features are precomputed and then cached, drastically reducing redundant computation. This paradigm shift reduces the number of alignment runs from N(N+1) to N for N monomers when screening all unique dimeric pairs.
Model Compilation and Bucketing
For AF2, inference throughput is further enhanced by grouping target pairs into sequence-length buckets and padding sequences within each bucket to a uniform length; JAX compilation then occurs only once per bucket. This dramatically decreases GPU occupation time for compilation, becoming especially beneficial for short sequences where compile time is otherwise a dominant cost. While AF3 natively implements generalized bucketing and padding, AF_Cache standardizes and automates their usage for the full interaction prediction task.
AF_Cache supports both the FASTA-based input mode of AF2 and the JSON-driven interface of AF3. It automates dataset handling, dependency management, and database provisioning within HPC and single-machine environments, streamlining deployment of complex AlphaFold-based screening pipelines.
Empirical Benchmarking and Numerical Results
Throughput Gains: MSA and Prediction Stages
The authors benchmarked AF_Cache on 5,050 unique human mitochondrial protein pairs, comparing against standard AF2.3 and AF3.0 pipelines.
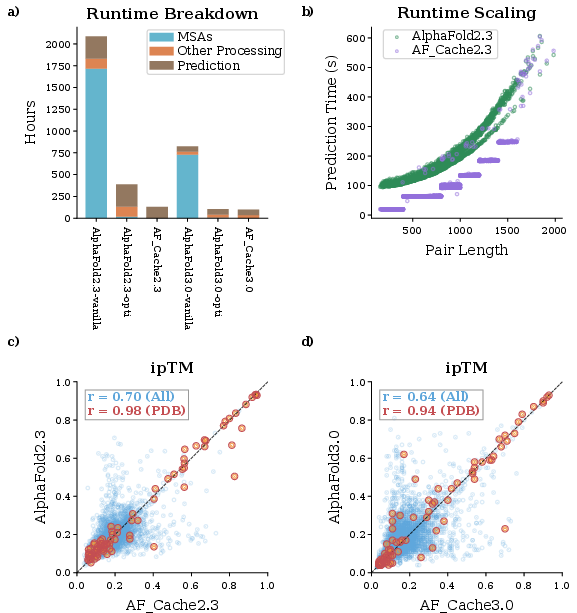
Figure 2: (A) Runtime decomposition reveals dominant acceleration of the MSA phase. (B) Prediction runtime scales identically with pair length, but AF_Cache is 90s faster per pair due to reduced JAX compilations. (C–D) ipTM score agreements demonstrate high structural congruence for structurally validated pairs.
- MSA Generation: GPU-accelerated and batched pipelines yield a 13-fold (AF2) and 5-fold (AF3) reduction in effective wall-time for MSA generation under a realistic core-parallelization scenario. In a raw core-hour comparison (not practical for real-world resource allocation), MSA runtime reduction can reach over three orders of magnitude.
- Prediction (Inference): For AF2, aggregate prediction and compilation time dropped from 253 to 125 GPU hours. The bucketing/padding strategy yields an average speedup of 90 seconds per protein pair, with compilation overhead virtually amortized across large buckets.
Structural Fidelity: ipTM Score Correlation
- Predicted interface TM-score (ipTM) values correlate robustly across different pipeline configurations. For pairs for which both monomers map to the same PDB entry, ipTM correlations exceed r≥0.94 (Pearson coefficient), indicating that structural predictions for pairs with experimental templates are highly consistent irrespective of the pipeline used.
- Across the entire dataset, ipTM agreement is more moderate (r=0.64–0.70), consistent with prior observations and reflecting both methodological stochasticity and intrinsic predictor uncertainty for weak coevolution and remote homology cases.
Comparative Analysis: AlphaFast, ColabFold, and AF_Cache
AlphaFast and ColabFold both introduced GPU-accelerated MSA generation and simplified batch orchestration for AlphaFold. AF_Cache extends these ideas in the following ways:
- Generates all unique monomeric MSAs upfront at workflow initialization, thus maximizing caching efficiency.
- Parallelizes database-specific MSA steps between CPU and GPU to further eliminate resource underutilization.
- Implements explicit bucketing and padding for dimeric and multimeric predictions in AF2, closing a feature gap with AF3.
- Integrates transparent support for pipeline parallelization across HPC clusters, supporting both AF2 and AF3, and enabling multi-node scaling.
Implications and Future Prospects
AF_Cache enables practical deployment of high-throughput, all-against-all PPI structural screening workflows based on AF2 and AF3 for large datasets, with computational cost reduced enough to render proteome-scale applications feasible. The high correlation among scores for PDB-supported interfaces suggests that predictive signal is retained while computation is drastically reduced. Moderate correlation for novel pairs highlights the persistent limitations of template-free prediction, but does not degrade relative to standard pipelines.
Given the modular structure of AF_Cache, integrating further speedups—for example, via large-scale distributed inference frameworks or prefiltering strategies using sequence features or low-resolution predictors—appears straightforward. As AF3 and future AlphaFold iterations continue to generalize to multicomponent biomolecular assemblies, caching and batching paradigms exemplified by AF_Cache will likely become the standard foundation for scalable structure-based interactomics.
Conclusion
AF_Cache presents a comprehensive, workflow-based system that removes recurring computational bottlenecks in large-scale AlphaFold-based protein–protein interaction prediction. By leveraging GPU-batched MSA, monomer feature caching, and intelligent input bucketing, it achieves substantial speedup with no compromise to the accuracy of structural predictions for empirically validated pairs. As the landscape of structure-based protein network biology expands, such scalable pipelines will be indispensable for both basic research and applied bioinformatics (2606.04566).

