- The paper introduces an O(n log log n)-time, O(n)-space algorithm, significantly reducing previous O(n log^3 n) time bounds for anchored edit distance chaining.
- It combines advanced techniques, including fast integer sorting and specialized owner-list data structures, to efficiently manage gap and overlap cost models.
- Experimental results demonstrate that the proposed llchain variant achieves up to 10× faster worst-case performance and linear scaling, making it practical for genomic alignment.
Revisiting O(nloglogn) Chaining for Anchored Edit Distance
Introduction and Context
This work addresses the computational complexity of colinear chaining under the "anchored edit distance" model, a critical component of modern sequence alignment pipelines, especially in genomics. The significance of anchored edit distance arises from its function as an efficiently-computable proxy for classical edit distance, with applications in mapping long reads to references where seeds (anchors) correspond to exact matches and further alignment extends through chaining.
Previous approaches, notably those by Jain, Gibney, and Thankachan, established an O(nlog3n) algorithmic upper bound for the anchored edit distance, combining specific gap and overlap cost models. This paper closes the gap between heuristics and theory, presenting an O(nloglogn)-time and O(n)-space algorithm matching the best complexities of early chaining results, but extended to the relevant cost model for modern applications.
Let T and Q be the sequences, and A the set of n exact match anchors (MEMs, MUMs, or fixed-length seeds). The chaining model allows overlaps between anchors, and the cost $\connect(a, a')$ of chaining a to O(nlog3n)0 is
O(nlog3n)1
where O(nlog3n)2 is the maximum gap, and O(nlog3n)3 is the diagonal distance. The total cost is minimized over all valid chains, with strong connections established between this and the edit-distance in alignments where only anchor matches are free.
Algorithmic Contributions
The paper presents an O(nlog3n)4-time, O(nlog3n)5-space solution by fusing several algorithmic designs:
- The O(nlog3n)6 gap cost handling of Eppstein et al. and Chao-Miller,
- The overlapping cost scheme of Baker and Giancarlo,
- Fast integer sorting and predecessor data structures for dynamic programming recurrence acceleration.
The optimal substructure is split into four exhaustive cases (overlap/gap, O(nlog3n)7/O(nlog3n)8 dominance), permitting specialized data structures per subproblem. For each anchor O(nlog3n)9, predecessors are found via line sweeps and efficient RMQ structures, with the most challenging scenario (gap-gap with O(nloglogn)0-dominance) handled by a compact dynamic owner-list (Figure 1) representing the region of influence efficiently.
Figure 2: Optimal recursive values for case 3 O(nloglogn)1, visualizing the owner-region partition central to linear-space, nearly-linear-time chaining.
The crux is that all possible predecessor queries per anchor can be supported in O(nloglogn)2 amortized time with only O(nloglogn)3 space, generalizing classic sparse dynamic programming to modern, overlap-permitting cost functions.
A practical O(nloglogn)4 variant, llchain, removes some complexity by employing standard balanced tree predecessors rather than word-RAM/van Emde Boas/Y-fast trie schemes, still yielding competitive performance on realistic genomics inputs.
Experimental Results
Empirical studies use PacBio HiFi reads, mapping them to the human genome. After seeding (MEM finding), anchors are chained using ChainX (heuristic), ChainX-opt (exact), and the new llchain (O(nloglogn)5).
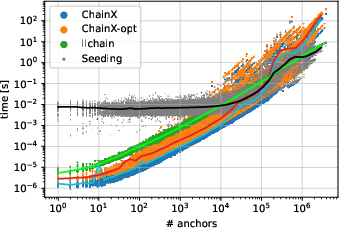
Figure 4: Time distribution for seeding and chaining with ChainX, ChainX-opt, and llchain on human genome MEMs shows llchain's low variance and robust scaling.
For abundant anchors (O(nloglogn)6 per query), ChainX and ChainX-opt's runtime grows superlinearly with broad variance due to seed distribution, often dominated by repetitive regions. In contrast, llchain's runtime grows linearly and is only marginally higher than the seeding cost itself. On average, llchain is O(nloglogn)7 faster in worst-case instances and O(nloglogn)8 faster overall compared to alternatives. Furthermore, the method provides a deterministic upper bound on computation time, avoiding adverse performance in large or repetitive input regions.
Theoretical and Practical Implications
This result demonstrates that the optimal chaining model underpinning anchored edit distance admits efficient computation in both theory and practice, closing a gap between best heuristics and known upper bounds. The techniques introduced—especially the compact owner-list for managing recursive subregions—extend the classical DP paradigm, enabling efficient handling of overlaps as well as complex cost metrics beyond simple linear/concave/convex formulations.
Practically, this enables deployment in long-read mapping, whole-genome alignment, large-scale assembly evaluation, and as a subroutine in advanced heuristics like A*-seeded path finding, where a chaining lower bound greatly accelerates subsequent extension.
On the theoretical side, the methodological advances may be translatable to broader classes of sparse dynamic programming problems, especially those with overlaps and asymmetric or mixed costs—a recurring pattern in modern sequence analysis.
Future Prospects
Potential extensions include gap-cost models required by the A*PA framework or alignment with inversions, and adaptation to arbitrary fragment scores, affine gap penalties, and inexact matches. Implementation improvements to leverage more advanced predecessor structures (e.g., B-trees or word-level parallelism) could further reduce constant overheads.
Conclusion
The work achieves optimal time (up to O(nloglogn)9) and space bounds for anchored edit distance computation via chaining, with practical implementations demonstrating strong empirical performance and low variance on realistic genomic datasets. By connecting and improving classical sparse dynamic programming, gap/overlap cost handling, and modern needs, the paper reinforces the relevance of algorithmic foundations for state-of-the-art computational genomics.