- The paper demonstrates that temporal graph neural networks, integrated with pseudotime-resolved snapshots, robustly forecast the evolution of gene regulatory networks.
- It uses a pipeline combining single-cell transcriptomics, SCENIC-derived GRNs, and TGNNs to achieve superior performance in gene-expression, link, and centrality forecasting.
- Empirical benchmarks reveal that spectral GNNs like GCRN-GRU and ChebNet capture regulatory hub dynamics more effectively than large-scale foundation models.
Temporal Graph Learning for Dynamic Gene Regulatory Networks: A Framework for Modeling Biological System Evolution
Introduction
The paper "Applications of temporal graph learning for predicting the dynamics of biological systems" (2605.28659) systematically evaluates the utility of temporal graph neural networks (TGNNs) for modeling and forecasting the evolution of biological systems. Emphasis is placed on the dynamics of gene regulatory networks (GRNs), derived from high-dimensional single-cell transcriptome data, and the explicit integration of temporal structure via pseudotime-resolved graph snapshots. The paper asserts that explicit modeling of regulatory structure, as opposed to static or sequence-based foundation model approaches, is critical for capturing nontrivial biological dynamics—an assumption validated empirically through a series of prediction tasks.
Temporal Graph Learning Pipeline for Developmental Biology
The methodological pipeline begins with single-cell transcriptomics, processed using diffusion pseudotime (DPT) estimation, which orders cells along developmental trajectories. Cells are discretized into temporally coherent bins, from which GRN snapshots are inferred using the SCENIC algorithm. These temporally indexed graphs—nodes as genes, directed edges as regulatory interactions—compose a sequence (G(1),...,G(T)). Each graph’s node features include aggregated gene expression statistics; edge features capture regulatory confidence scores.
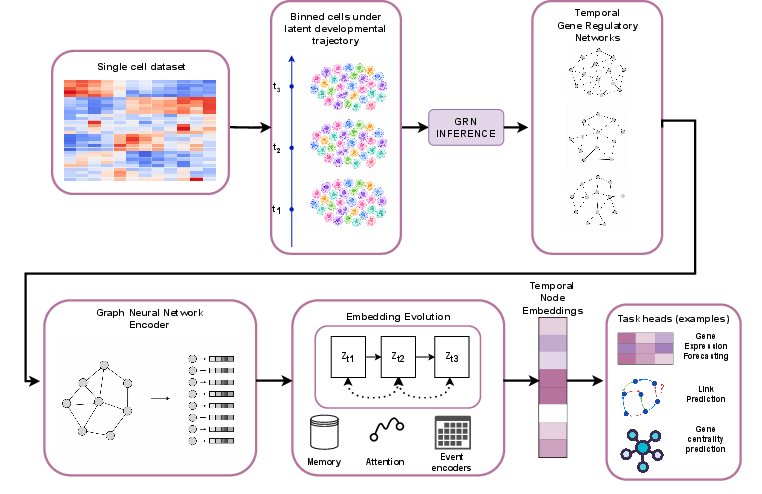
Figure 1: Overview of the proposed temporal graph learning pipeline for modeling and predicting the evolution of biological systems.
TGNNs are then applied, optimizing temporal representations of genes in the evolving regulatory context. The learned embeddings facilitate three downstream forecasting tasks: gene-expression change prediction, future regulatory link prediction, and future centrality projection.
The following tasks form the core evaluation benchmarks:
1. Gene-Expression Change Forecasting:
Given GRN snapshots up to time t, models predict the variation in mean gene expression between t and t+1 for each gene. This formulation is directly pertinent to evaluating dynamic aspects of virtual cell models and is compared against transformer-based biological foundation models (e.g., scGPT, scFoundation).
2. Future Link Prediction:
Models estimate the probability of a regulatory relationship between two genes in G(t+1) given the preceding GRN sequence. Edges are partitioned to increase prediction difficulty by sampling negatives among active (but unconnected) genes in a temporal snapshot, aligning with current best practices in dynamic link prediction evaluation.
3. Future Centrality Forecasting:
To gauge models’ capacity to anticipate changing network roles, future out-degree centrality—proportional to the number of regulatory targets per gene—is predicted at t+1. Central genes often correspond to lineage-determining transcriptional hubs.
The experimental setting is a live prequential framework, where models predict on the next temporal snapshot before being updated on it, reflecting a realistic use case and accommodating the evolving structure of biological systems.
Benchmarking and Numerical Insights
Multiple architectures—static GNNs, discrete-time TGNNs, supervised tabular models, and foundation models—were evaluated on two mouse developmental scRNA-seq datasets: erythroid gastrulation and pancreatic endocrinogenesis. Key observations include:
- Gene-Expression Forecasting:
TGNNs systematically outperform foundation models despite the latter being pretrained on orders of magnitude more data. Notably, GCRN-GRU and ChebNet architectures deliver the highest Pearson correlation coefficients. The performance advantage of spectral graph convolutions underscores the importance of multi-hop information flow in heterophilous, hub-centric GRNs. The gap between ChebNet and GCRN-GRU is consistently small, suggesting local regularity (Markovianity) in the evolution of GRNs.
Temporal models excel; GCRN-GRU attains the top AUPRCs on both datasets. Static memorization baselines (EdgeBank) perform strongly due to high edge recurrence but are surpassed by temporal GNNs, especially in later snapshots where the regulatory structure stabilizes but local rewirings remain nontrivial.
- Centrality Prediction and Regulatory Hub Dynamics:
Spectral GNNs, particularly ChebNet and GCRN-GRU, achieve superior correlation and precision in forecasting out-degree centrality. Temporal heatmaps extracted from GCRN-GRU’s predictions of erythroid gene-hub dynamics reveal biologically coherent shifts—early stages marked by biosynthetic/proliferative regulators, late stages dominated by canonical hematopoietic/erythroid transcription factors such as Klf1, Spi1, Erg, and Meis1.
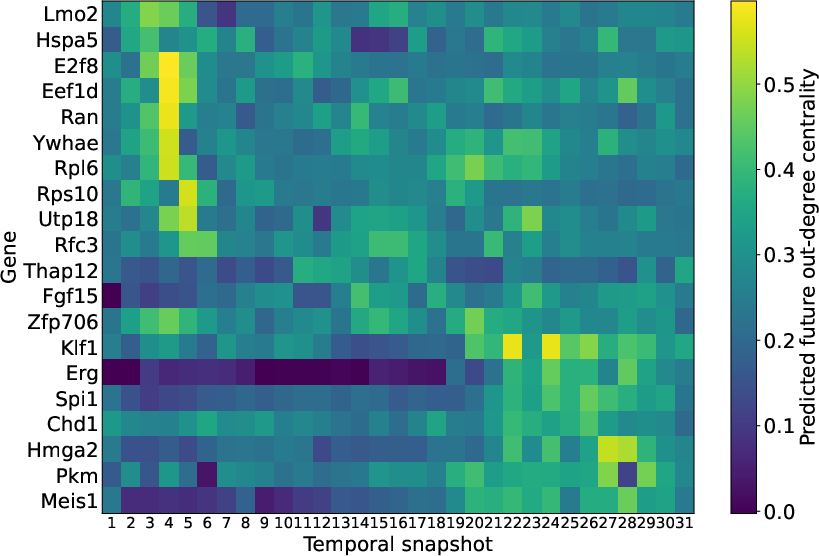
Figure 2: Temporal heatmap of the predicted future out-degree centrality for the top regulatory hubs identified by GCRN-GRU on mouse-gastrulation dataset.
The capacity to track and forecast regulatory hub emergence over developmental time demonstrates the analytical depth enabled by temporal graph learning, which is not possible using purely tabular, sequence-based, or static models.
Implications and Perspectives
The paper presents a contradictory empirical result: foundation models—despite being computationally intensive and trained on massive data with expressive transformer architectures—are outperformed on dynamic prediction tasks by temporally structured, graph-based models trained on limited data. This suggests that inductive bias toward temporal relational structure is more critical in dynamic biological forecasting than scale or general contextual embedding capacity.
Practically, the demonstrated pipeline offers a blueprint for in silico cell fate simulation, perturbation-response analysis, and identification of transiently central regulatory programs—capabilities central to regenerative medicine, disease progression modeling, and cellular engineering. The approach resolves the challenge of destructive single-cell measurement by reconstructing temporal progression via computation.
Theoretically, the finding that spectral GNN paradigms excel on dynamic, highly connected, heterophilous biological networks challenges the dominant focus on message passing in GNN design. It suggests a need to further investigate spectral methods and their correspondence with real-world long-range dependency management, particularly in hub-centric biological systems [arnaiz-rodriguez2026oversmoothing].
It is anticipated that further research will expand the benchmark to additional foundation models and introduce event-based temporal attention architectures (e.g., DyGFormer) to assess whether explicit temporal attention yields further improvements, especially as more granular developmental processes are targeted.
Conclusion
This study operationalizes a robust methodology for representing, modeling, and forecasting biological system dynamics with temporal GNNs. Empirical results establish the superiority of temporal graph-based approaches over static foundation models for a variety of forecasting tasks in developmental biology. The explicit encoding of regulatory structure and temporal evolution enables not only accurate prediction but also interpretable discovery of regulatory regime shifts and hub dynamics—benchmarking a new standard for dynamic biological modeling. The work motivates future integration of task-specific inductive biases and interpretable graph learning paradigms in computational biology, and opens directions for foundational advances in dynamic graph learning for complex, real-world systems.