PhylaFlow: Hybrid Flow Matching in Billera-Holmes-Vogtmann Tree Space for Phylogenetic Inference
Abstract: Phylogenetic trees are hybrid objects: branch lengths vary continuously, while topologies change discretely through edge contractions and expansions. Billera-Holmes-Vogtmann (BHV) tree space provides a canonical geometry for this structure, representing each resolved topology as a Euclidean orthant and topological changes as motion across shared lower-dimensional boundaries. We introduce PhylaFlow, a hybrid flow-matching model that learns posterior-basin transport in BHV tree space. PhylaFlow is trained on BHV geodesic paths from random starting trees to short-run posterior samples, coupling continuous branch-length motion within orthants with learned boundary events and discrete topology transitions. We evaluate the learned geometry operationally: if the flow reaches posterior-relevant regions, finite-budget Bayesian refinement initialized from, or guided by, its terminal trees should recover posterior-supported topologies more efficiently. Across DS1-DS8 phylogenetic posterior benchmarks, PhylaFlow substantially reduces initial Tree-KL relative to classical initializers. After finite-budget MrBayes refinement, direct PhylaFlow improves early and intermediate topology-recovery trajectories on most datasets, while split-guided PhylaFlow-MCMC obtains the strongest hard-case results. The best PhylaFlow variant outperforms short-warmup on seven of eight datasets and PhyloGFN on five of eight under the same refinement budget. In a joint sequence-conditioned experiment, sequence embeddings steer posterior split recovery, although exact posterior topology recovery remains preliminary. These results show that hybrid flow matching can learn actionable transport in BHV tree space and provide a geometry-aware proposal mechanism for Bayesian phylogenetic inference.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
Explaining “PhylaFlow: Hybrid Flow Matching in BHV Tree Space for Phylogenetic Inference”
What is this paper about?
This paper introduces PhylaFlow, a new AI method to help scientists figure out “family trees” of species, viruses, or genes (called phylogenetic trees). These trees show how things are related and how they evolved over time. Instead of guessing random trees and slowly fixing them, PhylaFlow gives smarter starting points that are already closer to the right answers, so standard tools can finish the job faster.
What questions are the authors trying to answer?
The authors tackle a practical question: can a neural network learn how to “move” through the space of all possible trees in a smart way—so it quickly gets near the regions that the data supports? In simpler terms: can we teach an AI to navigate a huge maze of possible evolutionary trees and drop us off close to the best parts of the maze?
They focus on:
- Learning how to change both branch lengths (how “long” an evolutionary step is) and the tree’s shape (who is grouped with whom).
- Testing whether these smart starts actually help a popular Bayesian method, MrBayes, find good trees faster and more reliably.
How does PhylaFlow work? (With simple analogies)
Imagine the “space of all trees” as a giant building made of rooms and doors:
- Each room is a specific tree shape (who is connected to whom).
- Inside a room, you can move furniture around (change branch lengths) freely.
- To switch to a different tree shape, you go to the door (a boundary), pass through, and enter a new room.
This building is called BHV tree space (Billera–Holmes–Vogtmann), a special geometry that treats tree shapes and branch lengths together.
PhylaFlow learns how to:
- Move smoothly within a room (adjust branch lengths),
- Decide which door to head toward (which branch should shrink to zero),
- And how to rebuild the connections when it passes through that door (change the tree shape).
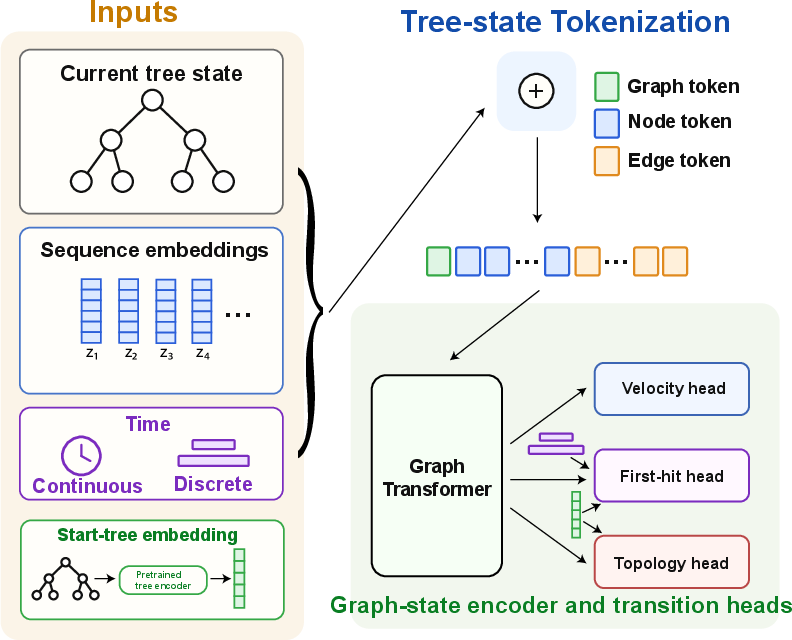
To make that happen, the model has three “heads” (sub-parts) that work together:
- Velocity head: tells how quickly each branch should grow or shrink (moving furniture in the room).
- First-hit head: predicts which branch will hit zero length first—the “door” to a new room.
- Topology head: decides how to reconnect the tree on the other side of the door (how the new room is arranged).
Training idea (simple version):
- The authors take “easy start” trees and “target” trees sampled from a rough-but-realistic posterior (think: a map of likely answers from a short MrBayes run).
- They compute shortest paths between these pairs in BHV space—like drawing a straight arrow through the building from start room to target room.
- PhylaFlow learns to imitate the direction and steps along these paths: how to move inside rooms, when to hit doors, and how to rebuild after crossing.
Two kinds of time:
- Within a room, time flows continuously (smooth branch changes).
- Each door crossing is a new phase (discrete steps).
After training, PhylaFlow takes any random start tree and “transports” it closer to likely regions—without using sequence likelihoods at that moment (no heavy calculations yet). Then MrBayes takes over to polish and fully calibrate the results.
How did they test PhylaFlow?
They used eight standard benchmark datasets (DS1–DS8). For each:
- They trained PhylaFlow using short MrBayes runs (to sketch what the “likely” tree regions look like).
- They then gave PhylaFlow unseen starting trees and compared:
- Initial quality (before any refinement),
- And quality after a fixed MrBayes budget (same number of steps for all methods).
They measured:
- Tree-KL: how close a set of proposed trees is to a long, high-quality reference posterior (lower is better). This checks if you’re in the right parts of the maze, not just at one lucky tree.
- Split-KL: a simpler check comparing how often particular “splits” (groups) appear.
- Likelihood gap: how plausible the generated trees are under the data (are they in the “high-scoring” zone?).
They also compared to strong baselines:
- Classic starts: random, maximum likelihood (IQ-TREE), maximum parsimony, UShER.
- Neural or posterior-informed methods: PhyloGFN (a strong likelihood-guided model) and a “short-warmup” baseline that starts directly from the same short MrBayes sketch used to train PhylaFlow.
Finally, they experimented with “conditioning” PhylaFlow on sequence embeddings (summaries of the input sequences) to steer it toward dataset-specific patterns.
What did they find and why is it important?
Main takeaways:
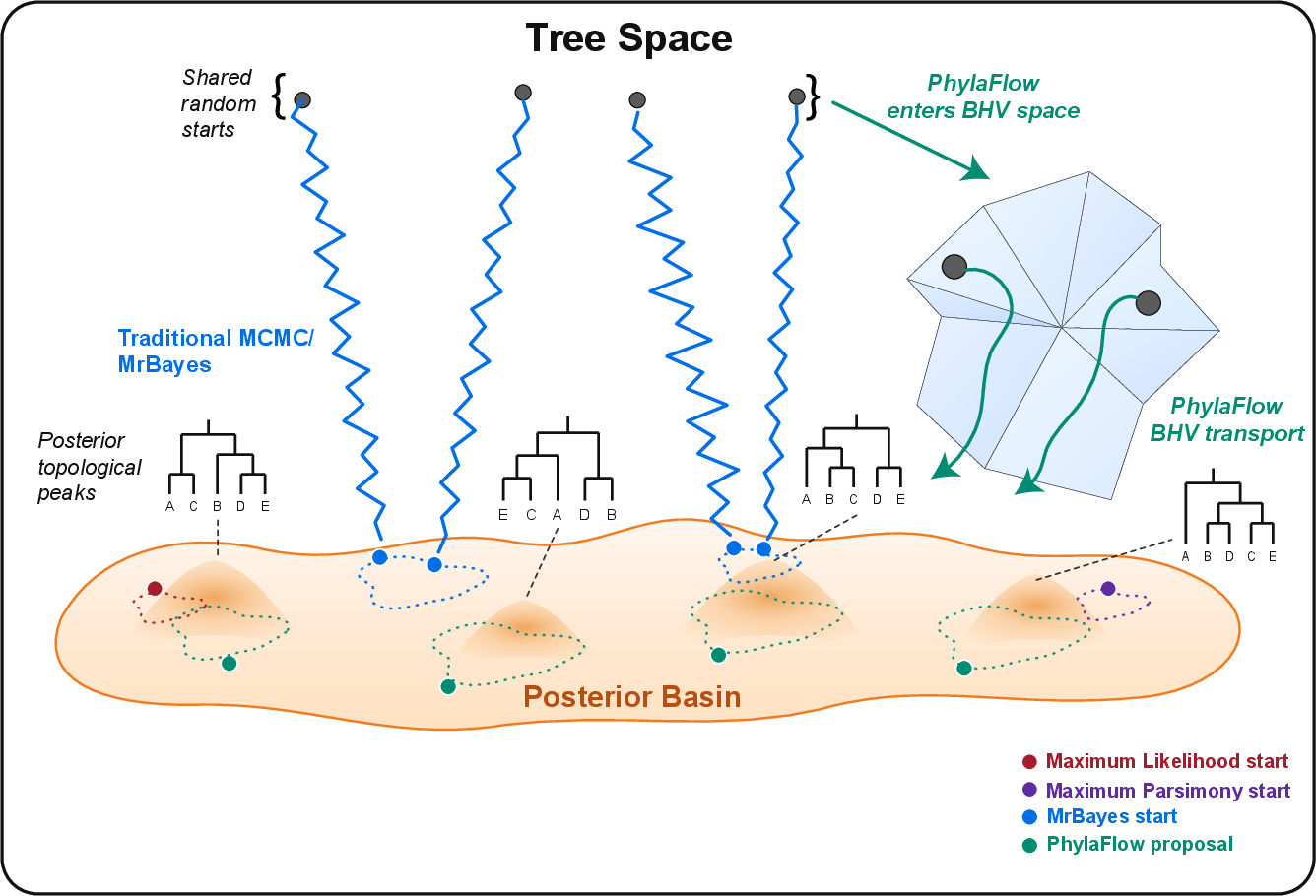
- Better starting points: Before any MCMC refinement, PhylaFlow’s proposed trees were much closer to the true posterior regions (best initial Tree-KL on all eight datasets). In other words, it drops you near the good neighborhoods of the maze right away.
- Faster early progress: With a fixed MrBayes budget, runs that started from PhylaFlow trees often reached better tree distributions earlier or midway through the run. This means less time burned “finding the right region.”
- Guided refinement helps on hard cases: A variant called PhylaFlow-MCMC used PhylaFlow’s learned split patterns to guide MrBayes moves. This gave the best results on several of the tougher datasets, even beating strong neural and posterior-informed baselines under the same compute budget.
- Plausible likelihoods: PhylaFlow’s starts weren’t “weird” or unlikely under the data. Their likelihood gaps were small or the best among initializers on most datasets. So it didn’t cut corners to get close in topology space—it kept trees plausible.
- Cost-quality balance: PhylaFlow delivered strong performance with lower setup time than some strong neural baselines (like PhyloGFN) on large datasets.
- Sequence conditioning shows promise: When given sequence embeddings, a single PhylaFlow model trained across multiple datasets could steer toward dataset-relevant splits (groupings), even if exact full-tree matching was still early-stage. A stress test showed the conditioning truly influenced outcomes.
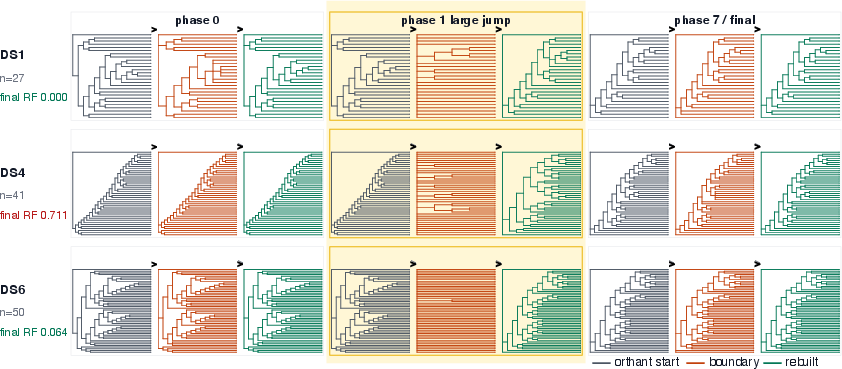
A neat behavioral detail:
- PhylaFlow often performs one big “collapse-and-rebuild” step first (a large move to a new topology), then makes smaller refinements—like making a big jump to the right neighborhood and then tidying the house.
Why does this matter?
- Phylogenetic inference is crucial for understanding how pathogens spread, how variants evolve, and how species are related. Faster, smarter starts can save time and compute, especially when many analyses need to be run quickly (e.g., in an outbreak).
- PhylaFlow doesn’t replace trusted Bayesian tools (like MrBayes). Instead, it works with them, getting you near the good areas sooner, so the Bayesian method can spend more time refining and less time wandering.
- The approach shows that geometry-aware learning (using the BHV “rooms and doors” view of tree space) can guide practical movement through a complex, hybrid space (part continuous, part discrete). That’s useful beyond this single application.
Limitations and what’s next
- Sampling speed: The topology update is step-by-step (autoregressive), which can be slow when many changes are needed.
- Generalization: A single model conditioned on sequences across many datasets is promising but not yet as strong as per-dataset training for exact topology recovery.
- Future directions: Faster sampling strategies, better sequence-to-tree training, and end-to-end learning that more tightly couples sequence signals with tree movement.
Bottom line
PhylaFlow learns how to navigate the “building of all trees” so it can deliver better starting points for Bayesian phylogenetic inference. This makes it easier and faster to find accurate evolutionary trees, especially when time and compute are limited.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a concise list of what remains missing, uncertain, or unexplored in the paper, phrased to guide follow-up research:
- Reliance on posterior-supervised training: How robust is PhylaFlow when the short-run posterior used for supervision is biased, poorly mixed, or obtained under misspecified models/priors? Evaluate sensitivity to different short-run chains, priors (Yule, birth–death, coalescent), and substitution models.
- Generalization beyond per-dataset training: The strongest results come from dataset-specific checkpoints; the jointly sequence-conditioned model underperforms on exact topology recovery. Can a single amortized model generalize across taxon sets, alignments, and evolutionary regimes without per-dataset retraining?
- Sequence conditioning is preliminary: Conditioning via frozen “Phyla” embeddings improves split-level recovery but not exact topology recovery. Do end-to-end learned sequence encoders, alignment-aware features, or site-pattern statistics provide stronger control and better topology-level accuracy?
- Scalability to large taxon counts: BHV dimension and the number of potential boundary events grow with taxa. How do training time, BHV geodesic supervision, and autoregressive boundary resolution scale to hundreds/thousands of taxa typical of surveillance datasets?
- Real-world complexity: Benchmarks (DS1–DS8) are small and standard. Performance remains untested on:
- Large, sparse, or noisy MSAs with indels and missing data
- Heterogeneous substitution processes (partitioned, codon, site-heterogeneous models)
- Molecular clock/time-calibrated (ultrametric) trees
- Recombination, gene-tree discordance, and species-tree inference
- Rapidly growing outbreak datasets (e.g., SARS-CoV-2–scale).
- Rooting and time inference: BHV formulation is unrooted; extensions to rooted tree spaces or ultrametric/time-tree BHV geometries are not addressed. How to incorporate clocks and temporal information into the hybrid flow?
- Alignment-free proposal vs. alignment-conditioned posterior: Inference-time transport ignores the MSA by design. For unseen datasets without per-dataset training, how much posterior signal can be captured without sequence conditioning? What is the trade-off between proposal quality and alignment-free inference?
- Diversity of proposals and basin coverage: Does PhylaFlow generate diverse proposals covering multiple posterior basins, or does it concentrate mass near one region (“mode collapse”)? Quantify proposal diversity and coverage vs. the reference posterior (e.g., via multi-basin occupancy and entropy).
- Interaction with MCMC kernels: Early improvements sometimes degrade by 100k generations, suggesting a mismatch with standard MrBayes moves. Which proposal kernels (e.g., informed NNI/SPR, path proposals, adaptive MH) best exploit BHV-informed starts while preserving correct calibration?
- Correctness and calibration of guided MCMC: The PhylaFlow-MCMC scheme biases local NNI moves using split weights. Are detailed balance and stationary distribution preserved (with appropriate MH corrections)? Provide proofs and diagnostics for posterior calibration under guidance.
- Training signal vs. true posterior geometry: Supervision uses BHV geodesics between starts and short-run posterior trees. Do BHV geodesic directions align with likelihood/posterior gradients? Would alternative training signals (e.g., likelihood-informed velocities, reinforcement learning, or score matching) yield better transport?
- Deterministic sampler and uncertainty: The sampling algorithm is deterministic. Can stochastic hybrid flows (e.g., stochastic flow matching or diffusion in BHV) better capture posterior multi-modality and proposal diversity?
- Boundary-event learning: The first-hit head and autoregressive topology head are trained to mimic geodesic-induced collapses/rebuilds. Are these topological transitions optimal for posterior access, or do alternative boundary policies outperform? Evaluate different collapse/rebuild strategies and non-autoregressive topology decoders.
- Start-tree distribution and case-conditioning: Training uses random starts with a frozen “metric encoder” for case conditioning. How sensitive are results to the distribution of start trees? Would training with ML/MP/UPGMA starts or mixture-of-starts improve robustness?
- Sensitivity to priors and model misspecification: Because training targets reflect the prior/model used in MrBayes short runs, how strongly do transport policies depend on those choices? Test cross-prior and cross-model transfer.
- Evaluation fidelity: Tree-KL and Split-KL are computed against a long-run MrBayes reference. How sensitive are conclusions to the reference’s convergence quality, finite-sample noise, and burn-in choices? Include convergence diagnostics and uncertainty intervals for Tree-KL/Split-KL estimates.
- Likelihood diagnostics: The “optimized-likelihood gap” uses likelihood-optimized topologies. Does this metric correlate with posterior mass across regimes and models? Explore alternative posterior-basin diagnostics that jointly consider topology and branch lengths.
- Computational cost bottlenecks: Training requires computing many BHV geodesics and autoregressive transitions; inference requires multiple model calls per boundary event. Investigate:
- Faster geodesic approximations or learned surrogates
- Batch/parallel boundary resolution
- Non-autoregressive topology generators for reduced latency.
- Integration with other Bayesian engines: The study centers on MrBayes. How do benefits translate to BEAST, RevBayes, or other samplers with different proposal kernels and priors (e.g., skyline birth–death, relaxed clocks)?
- Branch-length modeling and clocks: Branch lengths are learned as positive reals within orthants; no explicit handling of ultrametric constraints or rate variation is shown. How to incorporate clock models (strict/relaxed) and rate heterogeneity into transport while respecting BHV geometry?
- Taxon-set transfer: The joint model conditions on dataset embeddings but uses fixed taxon sets within each dataset. Can transport policies transfer to new taxa or partially overlapping taxon sets, possibly via permutation-equivariant architectures and tokenizations that handle taxon insertions/deletions?
- Robustness to poor supervision: If short-run posteriors are highly multi-modal or under-sampled, does PhylaFlow overfit spurious modes? Compare training with different short-run seeds and lengths; use posterior-tempering or aggregation to stabilize targets.
- Combining with variational/posterior samplers: How does PhylaFlow initialization compare or combine with VBPI/SBNs/ARTree or GFlowNets (e.g., hybrid initialization followed by variational refinement, or alternating flow-based proposals with GFlowNet exploration)?
- Theoretical guarantees: Provide formal analysis (or counterexamples) for when hybrid flow matching in BHV can reduce expected BHV distance or KL to the true posterior, and conditions under which learned transport is guaranteed to improve finite-budget MCMC efficiency.
- Broader phylogenetic settings: Extend and evaluate to:
- Species-tree inference under the multispecies coalescent (gene-tree heterogeneity)
- Ancestral state/trait inference integration
- Joint topology–alignment inference (co-estimation) where alignment uncertainty is large.
Practical Applications
Practical Applications of PhylaFlow: Hybrid Flow Matching in BHV Tree Space
PhylaFlow is a geometry-aware, hybrid neural transport model that learns to move phylogenetic trees through Billera–Holmes–Vogtmann (BHV) tree space toward posterior-relevant basins, improving early and intermediate stages of Bayesian phylogenetic inference. It couples continuous branch-length flows with learned discrete topology transitions, and can be used as (i) a drop-in initializer for samplers like MrBayes and (ii) a split-guided proposal mechanism (PhylaFlow-MCMC). Below are specific, real-world applications mapped to sectors, with feasibility and dependencies noted.
Immediate Applications
The following can be deployed now using the released code and standard Bayesian phylogenetics tools.
- Accelerated Bayesian phylogenetic inference via smarter initialization (academia, healthcare, biotech)
- What: Use PhylaFlow to initialize MrBayes/BEAST runs from proposals closer to posterior basins, reducing burn-in and improving early topology recovery under fixed budgets.
- Tools/workflows: “PhylaFlow → MrBayes/BEAST” pipeline; Nextflow/Snakemake module that generates k diverse PhylaFlow starts per dataset and launches parallel chains.
- Assumptions/dependencies: Requires an MSA, substitution model, and priors for downstream MCMC; performance validated on DS1–DS8 (moderate-size benchmarks); BHV geodesic computation and GPU for model inference; generalization to much larger taxon sets may require tuning.
- Split-guided MCMC to improve mixing in hard posteriors (academia, healthcare)
- What: Use PhylaFlow-MCMC (split-weight guidance from PhylaFlow’s proposals) to bias local moves (e.g., NNI) toward posterior-relevant splits, improving topology-distribution agreement under finite budgets.
- Tools/workflows: PhylaFlow-generated guide bank → split weights → modified MCMC proposals (e.g., wrapper around MrBayes or a companion utility).
- Assumptions/dependencies: Requires integration layer to pass split weights to the sampler; improvements depend on posterior ruggedness; verify with Tree-KL and split-KL diagnostics.
- Real-time genomic surveillance triage and early situational awareness (public health policy, healthcare)
- What: Before long runs complete, use PhylaFlow to quickly propose candidate topologies and assess early convergence (Tree-KL/likelihood gap), providing faster guidance on transmission clusters/lineages (e.g., SARS-CoV-2, TB).
- Tools/workflows: Surveillance pipeline step that pre-screens datasets with PhylaFlow; dashboard that reports early split recovery and likelihood plausibility.
- Assumptions/dependencies: Requires validated operating procedures and thresholds for early-use decisions; regulatory environments may require confirmatory analyses.
- Compute-cost reduction and throughput increase in phylogenetics pipelines (industry, academia)
- What: Reduce the number of MCMC generations and/or number of random restarts by seeding with PhylaFlow proposals, increasing throughput on shared HPC/cloud resources.
- Tools/workflows: Scheduler-aware batch generation of PhylaFlow starts; pipeline knobs for budget reallocation based on early Tree-KL.
- Assumptions/dependencies: Gains are dataset-dependent; requires monitoring infrastructure for convergence diagnostics.
- Quality control for model/ alignment issues via likelihood-gap checks (academia, industry)
- What: Use the optimized-likelihood gap diagnostic on PhylaFlow’s proposed trees to detect datasets that are far from plausible likelihood regions (flagging potential alignment/model misspecification).
- Tools/workflows: Automated QC step after PhylaFlow generation that computes a small set of optimized likelihoods and thresholds “gap” alerts.
- Assumptions/dependencies: Requires fast likelihood optimization on proposed topologies (e.g., IQ-TREE/RAxML optimization mode); thresholds must be calibrated per model class.
- Diversity-aware start-tree banks for robust inference (academia, healthcare)
- What: Generate a diverse set of basin-entry proposals (multiple modes) to reduce start-point bias and improve coverage of posterior modes in multi-run protocols.
- Tools/workflows: Bank-of-starts generator that enforces diversity via BHV or RF distance thresholds; feed multiple chains with non-redundant starts.
- Assumptions/dependencies: Requires diversity metrics and stopping rules; tractable on moderate-size datasets.
- Teaching and visualization of tree-space geometry (education, daily life)
- What: Use PhylaFlow’s trajectories to illustrate BHV motion (branch-length flows, boundary crossings) in courses/outreach, making phylogenetic inference more intuitive.
- Tools/workflows: Classroom notebooks and web demos that animate PhylaFlow phases and compare to geodesics/MCMC.
- Assumptions/dependencies: Requires simple wrappers for visualization and curated toy datasets.
- Open-source integration modules (software)
- What: Package PhylaFlow as a plugin/wrapper for common tools (MrBayes, BEAST, RevBayes) and as a Snakemake/Nextflow process, lowering adoption barriers.
- Tools/workflows: Containerized PhylaFlow with CLI; pipeline recipes that accept FASTA/MSA and output guided initial states or split weights.
- Assumptions/dependencies: Maintenance and version compatibility with evolving samplers; testing on user datasets.
Long-Term Applications
These require further research, scaling, or productization beyond the paper’s current scope.
- Amortized, sequence-conditioned phylogenetics for near–real-time inference (healthcare, industry)
- What: A single model that conditions on sequence embeddings to propose dataset-specific trees across taxon sets, reducing dependence on long MCMC runs for routine analyses.
- Potential products: “PhyloAccel” SaaS/API that returns proposal banks and split weights on upload; edge-accelerated modules for outbreak response.
- Assumptions/dependencies: Stronger cross-dataset generalization than shown; end-to-end training with embeddings; calibration layers to avoid bias; validation for regulatory use.
- BHV-aware proposal kernels in major samplers (software, academia)
- What: Incorporate learned boundary events and topology transitions directly as reversible proposals in MrBayes/BEAST/RevBayes to improve mixing and exploration of multi-basin posteriors.
- Potential tools: New reversible-jump moves informed by PhylaFlow’s first-hit and topology heads; plug-ins exposing BHV-geodesic structure.
- Assumptions/dependencies: Detailed balance and acceptance-ratio derivations; rigorous validation; community acceptance and maintenance.
- Scaling to very large trees and high-throughput settings (software, healthcare)
- What: Non-autoregressive or batched topology updates, better parallelization, and approximate BHV computations to handle thousands–tens of thousands of taxa.
- Potential tools: GPU-accelerated BHV libraries; sparse/low-rank approximations; streaming inference for incremental datasets.
- Assumptions/dependencies: Algorithmic advances for BHV geodesics and structured-subset decoding; memory efficiency; benchmark suites for large-scale evaluation.
- Integration with multi-gene/species-tree pipelines (academia, biotech)
- What: Use PhylaFlow to propose gene trees or species trees under coalescent models (e.g., MSC), improving initialization for hierarchical Bayesian frameworks.
- Potential tools: Wrappers for BPP/StarBEAST-like workflows; banks of gene-tree proposals feeding species-tree inference.
- Assumptions/dependencies: Extension from single-tree BHV geometry to hierarchical settings; joint handling of discordance and model priors.
- Active sampling and experiment design in genomic surveillance (policy, healthcare)
- What: Use uncertainty in early split recovery to prioritize additional samples or genomic regions that reduce posterior ambiguity most efficiently.
- Potential tools: Acquisition functions over splits/clades; dashboards recommending targeted sequencing or contact tracing priorities.
- Assumptions/dependencies: Real-time data pipelines; ethical/regulatory frameworks for adaptive sampling; reliable uncertainty quantification.
- Cross-domain hybrid transport in structured spaces (software, robotics, EDA)
- What: Apply “hybrid flow in orthant-like spaces” to other combinatorial–continuous problems (e.g., robot motion with mode switches, circuit layout with discrete structures), inspired by BHV transport.
- Potential products: Libraries for hybrid flow matching on piecewise-Euclidean manifolds; proposal generators for combinatorial optimization.
- Assumptions/dependencies: Suitable geometric formulations for target domains; availability of supervised trajectories; domain-specific constraints.
- Clinical decision support for AMR lineage tracking (healthcare)
- What: Faster lineage assignment and uncertainty-aware phylogenies in hospital outbreaks (e.g., TB, MRSA), accelerating infection control decisions.
- Potential tools: EHR-integrated modules that surface early clade structure and confidence for infection prevention teams.
- Assumptions/dependencies: Clinical validation, regulatory compliance, data integration; SOPs for acting on early phylogenetic signals.
- End-to-end sequence-to-tree training (academia, industry)
- What: Jointly learn sequence embeddings and BHV transport to align representation learning with downstream phylogenetic transport.
- Potential tools: Co-trained encoders with supervision from posterior sketches and BHV geodesics; self-supervised pretraining on large genomic corpora.
- Assumptions/dependencies: Large, diverse training sets; careful handling of model misspecification; compute budget for co-training.
Notes on Feasibility and Dependencies
- Training supervision: Current workflow uses short-run MCMC sketches to derive targets (geodesic paths to posterior samples). This setup imposes an upfront compute cost and depends on the chosen evolutionary model and priors.
- Generalization: Demonstrated on benchmark datasets (DS1–DS8). Real-world performance may vary with taxon count, rate heterogeneity, and dataset complexity.
- BHV infrastructure: High-quality BHV geodesic computation and utilities are needed for training and analysis; further optimization may be required for large-scale use.
- Integration: Most high-impact applications assume tight integration with established samplers (MrBayes, BEAST, RevBayes) or likelihood optimizers (IQ-TREE/RAxML).
- Statistical guarantees: PhylaFlow proposals complement, not replace, calibrated Bayesian inference. Final scientific or policy decisions should rely on posterior-calibrated results.
Glossary
- Amortized inference: A technique that learns a reusable mapping to approximate a posterior, enabling fast inference on new instances. "Recent machine-learning approaches instead use amortized or variational inference methods to approximate the posterior more efficiently"
- ARTree: An autoregressive neural model that parameterizes distributions over phylogenetic tree topologies. "Subsplit Bayesian networks (SBNs)~\citep{zhang2018sbn}, ARTree~\citep{xie2023artree}, and ARTreeFormer~\citep{xie2025artreeformer} parameterize increasingly flexible topology distributions"
- ARTreeFormer: A transformer-based autoregressive model for flexible distributions over tree topologies. "Subsplit Bayesian networks (SBNs)~\citep{zhang2018sbn}, ARTree~\citep{xie2023artree}, and ARTreeFormer~\citep{xie2025artreeformer} parameterize increasingly flexible topology distributions"
- Bayesian phylogenetic inference: The approach of inferring a posterior distribution over phylogenetic trees and branch lengths given sequence data and priors. "The goal of Bayesian phylogenetic inference is to characterize the posterior distribution over tree topologies and branch lengths given an observed sequence alignment,"
- BEAST: A Bayesian MCMC software package for phylogenetic analysis. "as in MrBayes and BEAST"
- BHV metric: The distance function defined on Billera–Holmes–Vogtmann tree space that enables geodesic computation between trees. "We review the formal BHV metric and geodesic computation in Appendix~\ref{Appendix:BHV}."
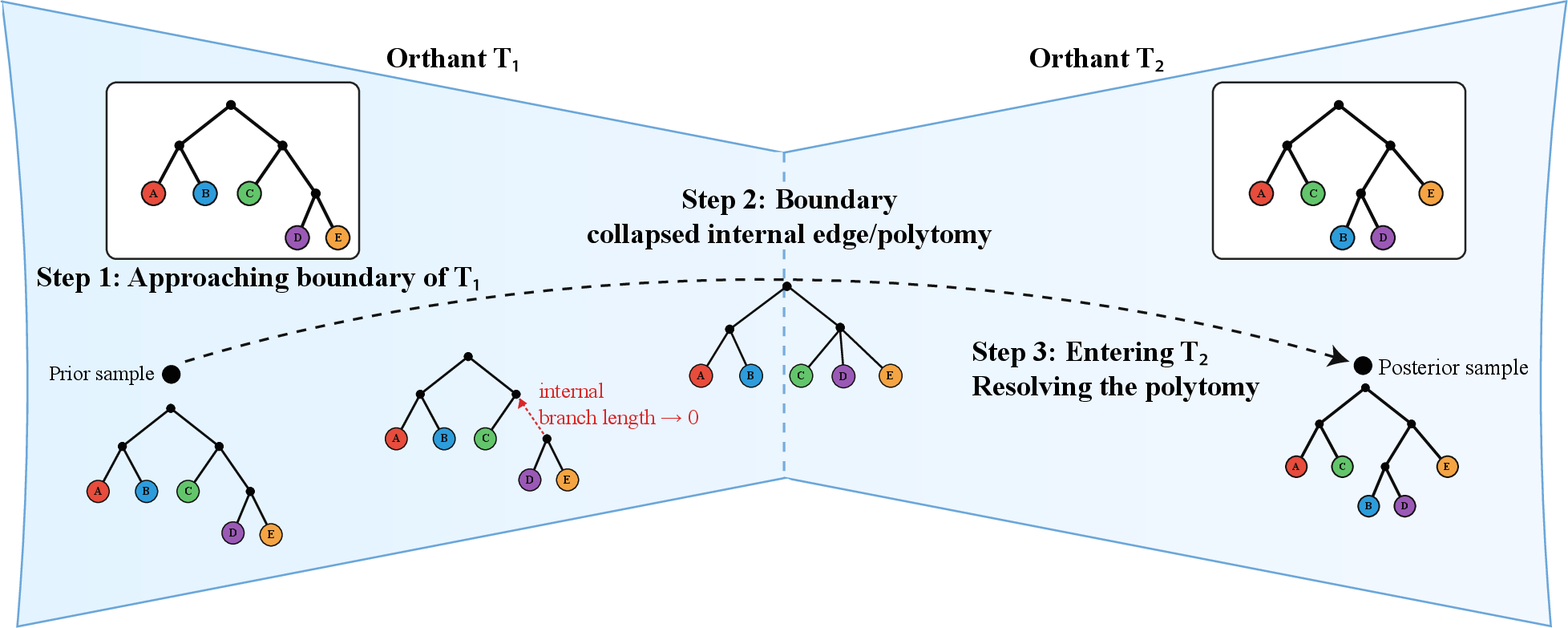
- BHV tree space: The Billera–Holmes–Vogtmann geometric space where each resolved topology is an orthant and topology changes correspond to crossing shared boundaries. "BilleraâHolmesâVogtmann (BHV) tree space provides a canonical geometry for this structure, representing each resolved topology as a Euclidean orthant and topological changes as motion across shared lower-dimensional boundaries."
- Branch lengths: Positive real-valued attributes on tree edges representing evolutionary time or expected substitutions. "Branch lengths vary continuously, while topological changes occur through edge contractions and expansions"
- Conditional flow matching: A training objective for learning velocity fields conditioned on inputs to realize transport between distributions. "which is trained by conditional flow matching on supervised branch-length velocities."
- Euclidean orthant: A nonnegative quadrant in Euclidean space; in BHV space, each orthant corresponds to a fixed tree topology with coordinates as internal branch lengths. "representing each resolved topology as a Euclidean orthant and topological changes as motion across shared lower-dimensional boundaries."
- Geodesic (BHV geodesic): The shortest path under the BHV metric connecting two trees in tree space. "PhylaFlow is trained on BHV geodesic paths from random starting trees to short-run posterior samples,"
- Geodesic velocity: The instantaneous velocity along the BHV geodesic used to supervise branch-length dynamics. "the velocity head is supervised with the geodesic velocity"
- GFlowNet: A generative flow network framework that learns policies to sample objects (here, trees) proportionally to a reward (e.g., likelihood-based). "it learns a GFlowNet policy that sequentially constructs trees"
- Marginal likelihood (model evidence): The probability of the data under the model, integrating over all trees and branch lengths; used for Bayesian model comparison. "The denominator is the marginal likelihood, or model evidence."
- Markov chain Monte Carlo (MCMC): A class of sampling algorithms for approximating posterior distributions by constructing a Markov chain whose stationary distribution is the target posterior. "classical Markov chain Monte Carlo (MCMC) methods may spend substantial finite compute"
- Maximum parsimony: A phylogenetic criterion that selects the tree minimizing the total number of character changes. "maximum parsimony starts"
- MrBayes: A widely used Bayesian MCMC sampler for phylogenetic inference. "MrBayes \cite{huelsenbeck_mrbayes_2001}, a widely used Bayesian phylogenetic MCMC sampler,"
- Multiple-sequence alignment (MSA): An alignment of homologous sequences across taxa used for likelihood-based phylogenetic inference. "After training, this proposal step does not require a multiple-sequence alignment (MSA)"
- Nearest-neighbor interchange (NNI): A local tree rearrangement move used in phylogenetic search and MCMC proposals. "uses these weights to bias local NNI proposals toward PhylaFlow-supported splits"
- Optimal transport: A mathematical framework for transporting one distribution to another with minimal cost; here used to conceptualize mapping start-tree distributions to posteriors. "Reframing Phylogenetics as an Optimal Transport Problem"
- Phylogenetic likelihood: The probability of the observed alignment given a tree, branch lengths, and a substitution model. "optimized against the phylogenetic likelihood and prior"
- PhyloGFN: A GFlowNet-based method that learns to sample phylogenetic trees guided by likelihood-derived rewards. "PhyloGFN is closer to posterior sampling: it learns a GFlowNet policy that sequentially constructs trees"
- Polytomy: A node in a tree where more than two lineages diverge, representing an unresolved branching order. "the collapsed tree may contain an unresolved polytomy."
- Posterior basin: A region of tree space containing high posterior mass; reaching it accelerates subsequent Bayesian refinement. "entering the posterior basin."
- Rate heterogeneity model: A model component capturing variation in substitution rates across sites or lineages. "such as the substitution model, rate heterogeneity model, or tree prior"
- Robinson–Foulds distance: A metric on tree topologies counting differing splits (bipartitions) between trees. "RF denotes Robinson--Foulds distance to the reference tree."
- Split-KL: A divergence that compares posterior split (bipartition) frequency distributions between methods and a reference posterior. "Split-KL provides a coarser marginal view by comparing posterior split frequencies."
- Subsplit Bayesian Networks (SBNs): Probabilistic models over tree topologies via subsplits, used for posterior approximation or density estimation. "Subsplit Bayesian networks (SBNs)~\citep{zhang2018sbn}, ARTree~\citep{xie2023artree}, and ARTreeFormer~\citep{xie2025artreeformer} parameterize increasingly flexible topology distributions"
- Substitution model: A probabilistic model specifying how sequence characters change along branches of a phylogenetic tree. "such as the substitution model, rate heterogeneity model, or tree prior"
- Tree-KL: A Kullback–Leibler divergence measuring disagreement between topology distributions from a method and a reference posterior. "We therefore measure Tree-KL, a divergence measure comparing sampled tree-topology distributions to a long-run reference posterior,"
- Tree prior: The prior distribution over tree topologies and branch lengths in Bayesian phylogenetics. "such as the substitution model, rate heterogeneity model, or tree prior"
- UShER: A parsimony-based tool for rapid phylogenetic placement and tree construction, used here as an initializer. "UShER-based initializers"
- Variational inference: An optimization-based approach that approximates a posterior by fitting a tractable family of distributions. "amortized or variational inference methods"
Collections
Sign up for free to add this paper to one or more collections.