- The paper introduces an exact density functional theory that integrates both symmetry-conserving and symmetry-breaking pair correlations to predict phase transitions with high accuracy.
- It employs harmonic expansion techniques and iterative Ornstein-Zernike solutions to analyze correlation functions in nematic and crystalline phases.
- The approach accurately delineates phase boundaries for fluid–solid and solid–solid transitions, matching simulation data for structural and elastic properties.
Ordering, Correlation Functions, and Phase Transitions in Molecular Systems: An Authoritative Review
Theoretical Foundations and Motivation
The paper "Ordering, correlation functions and phase transitions in molecular systems" (2605.18326) delivers a comprehensive synthesis of classical density functional theory (DFT) and its extension to systems with broken symmetry—phases such as crystals and liquid crystals. While DFT has long been established as a fundamental tool for homogeneous fluids, the treatment of phases where symmetry is broken (e.g., transition from isotropic fluid to crystalline solid or nematic) required significant advances both in conceptual framework and computational techniques. The paper extensively reviews the modern methodology for calculating pair correlation functions (PCFs) in ordered phases, their integration into DFT functionals, and their practical ramifications for phase transition analysis.
Classical DFT and its Extension to Broken Symmetry Phases
The basis of the formalism lies in representing the grand thermodynamic potential as a functional of the one-particle density. In homogeneous and isotropic fluids, PCFs are routinely obtained; however, in systems exhibiting spatial or orientational order, additional symmetry-breaking contributions to PCFs emerge. The direct correlation function (DCF) and the total pair correlation function are thus decomposed as:
c(x1,x2)=c(0)(x1,x2)+c(b)(x1,x2)
h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)
where superscripts (0) and (b) denote symmetry-conserving and symmetry-breaking components, respectively. This decomposition enables a systematic approach where the symmetry-conserving part is handled using integral equation theory (IET) adapted from fluid theory, and the symmetry-breaking part is constructed perturbatively in terms of the crystalline order parameters.
Order Parameters, Harmonic Expansions, and Broken Symmetries
A major structural innovation discussed is the harmonic expansion of densities and correlation functions in appropriate bases (Fourier for translation, spherical harmonics/Wigner D-matrices for orientation). This defines positional, orientational, and mixed order parameters, allowing precise characterization of transitions such as isotropic-nematic, nematic-smectic, and liquid-solid.
For example, in the nematic phase (uniaxial ordering), the molecular orientation distribution function is expanded in terms of spherical harmonics, and only certain Pl order parameters contribute, as dictated by the phase and molecular symmetry. For crystalline solids, the one-particle density is represented as overlapping Gaussians at lattice sites, with Fourier coefficients μG acting as order parameters.
Exact Density Functional Theory (EDFT) for Broken Symmetry Phases
The text progresses beyond traditional approaches such as the Ramakrishnan-Yussouff (RY) theory and weighted density approximations (WDA), which are insufficiently accurate for phases with strong order or soft interparticle potentials. Instead, it details an "exact DFT" (EDFT), in which the free-energy functionals explicitly integrate both symmetry-conserving and symmetry-breaking components of PCFs. The functional minimization problem thus becomes:
A[ρ]=Aid[ρ]−21∫ρ(x1)ρ(x2)cˉ(0)(x1,x2)−21∫ρ(b)(x1)ρ(b)(x2)cˉ(b)(x1,x2)
This explicit dependence on the broken symmetry part of the DCF is crucial, especially for soft potentials and subtle phase competitions (e.g., fcc vs. bcc).
Correlation Functions in Nematic Phases and Iso-Nematic Transition
The calculation of PCFs in the nematic phase is executed via iterative solutions of the Ornstein-Zernike equation, incorporating closure relations such as the mean spherical approximation (MSA) or Percus-Yevick (PY):
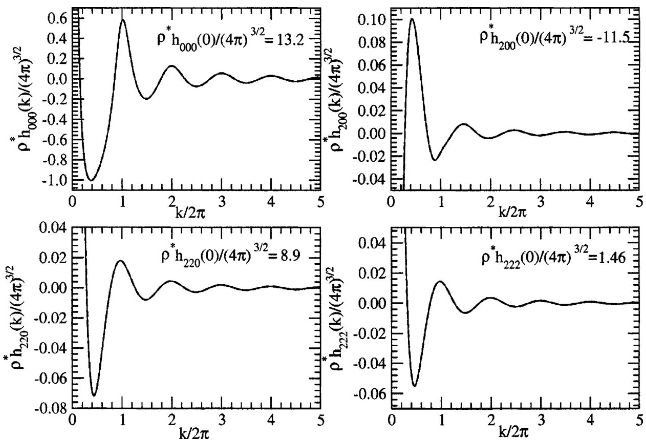
Figure 1: Fourier-space comparison of selective harmonic components of the total pair correlation function for the nematic phase under MSA, showing indistinguishable results between numerical and analytical methods.
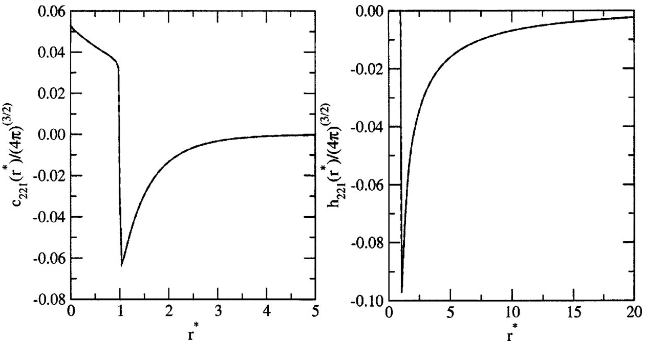
Figure 2: Analytical versus numerical comparison for harmonic coefficients relevant to nematic elasticity, exposing the characteristic 1/r∗ tail in h221(r∗) due to symmetry breaking.
The emergence of long-ranged tails (∼1/r) in certain harmonics of h is attributed to Goldstone modes associated with director fluctuations in nematics, with direct implications for elastic moduli and fluctuations.
The structure factor in the nematic is also shown to have a peak near k=0 due to enhanced effective attractions from parallel alignment. This precise treatment enables reliable identification of isotropic-nematic coexistence, with order parameters and densities in excellent accord with simulation:
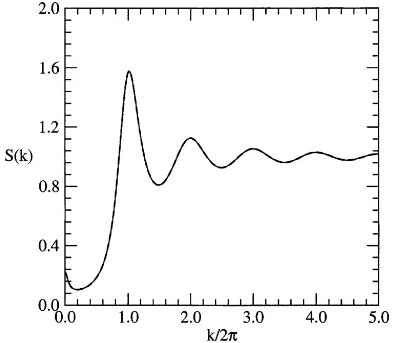
Figure 3: Overlapping analytical and numerical structure factor results for the nematic phase affirming the approach’s accuracy.
Crystalline Solids: PCFs, Higher-Order Direct Correlation Functions, and Series Convergence
For crystals, the expansion of h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)0 in powers of the density modulation h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)1 leads to diagrammatic series where higher-order direct correlation functions (e.g., h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)2, h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)3) are reconstructed from density derivatives of the two-body DCF and solved via methods akin to those of Barrat et al. The first term in this series is generally dominant as confirmed by comparative analysis of their Fourier transforms.
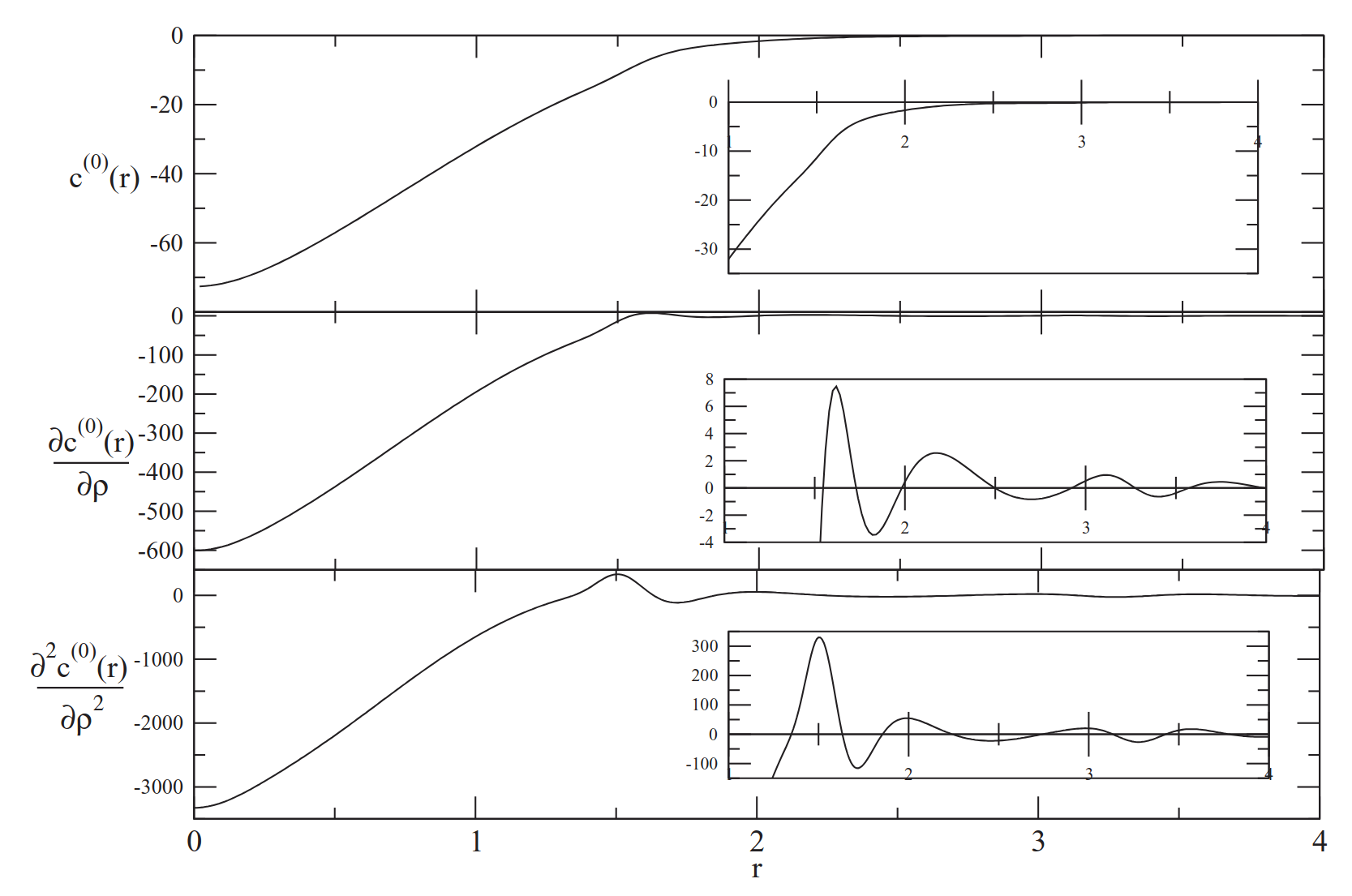
Figure 5: Variation of the free energy difference h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)4 as a function of localization and density jump parameters, pinpointing the coexistence minimum.
The approach yields crystallization parameters for inverse power-law potentials matching simulation for both fcc and bcc structures, including precise Lindemann ratios, coexistence pressures, and density jumps over a wide range of softness indices. The relative stability of close-packed and open structures (fcc/bcc) is predicted with strong fidelity.
Fluid–Solid and Solid–Solid Phase Boundaries
The EDFT formalism is further validated by mapping the phase boundaries (fluid–solid and bcc–fcc) for inverse power-law and Lennard-Jones-like potentials. The predicted triple points, phase coexistence regions, and critical softness parameter h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)5 for the transition agree quantitatively with large-scale simulations.
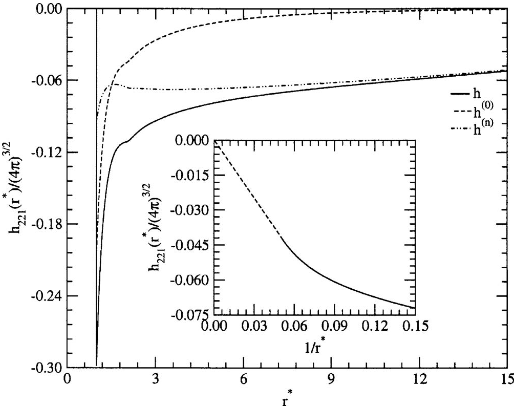
Figure 6: Phase diagram in (1/n, h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)6) space for systems with inverse power-law potentials, showing excellent correspondence between theoretical predictions (lines) and simulation data (symbols) for fluid–solid and bcc–fcc boundaries.
Additionally, the theory captures subtleties in two-dimensional crystals (e.g., soft disks with h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)7 interactions), with the calculated order parameters and correlation functions matching experiment and simulation even in the context of KTHNY-type transitions.
Implications, Impact, and Future Directions
Numerical Comparisons and Claims:
- The EDFT robustly predicts phase boundaries, coexistence densities, pressure, Lindemann ratios, and structural order parameters for both molecularly simple and complex systems.
- The broken symmetry contribution to the free energy, negligible for hard spheres, is demonstrated to contribute up to 45% for soft potentials (h(x1,x2)=h(0)(x1,x2)+h(b)(x1,x2)8), and omitting it grossly overestimates fluid stability.
- The approach enables accurate location of solid–solid transitions, including subtle bcc-fcc boundaries—an achievement beyond prior SODFT or WDA functionals.
Contradiction of Previous Approximations:
- Approximations such as RY/SODFT severely underestimate the stability region for solids and mispredict structural stabilities in soft potentials.
- Inclusion of explicit symmetry-breaking PCF contributions is essential even for qualitative correctness in many cases.
Theoretical and Practical Implications:
- The formalism unifies the treatment of both orientational and translational symmetry breaking, delivering a single prescription for all phase transitions in molecular systems.
- By reconstructing higher-order DCFs systematically from lower-order density derivatives, it may serve as a bridge to incorporate complex fluctuation phenomena (defects, elasticity, low-dimensional ordering, etc.).
- The flexible expansion in harmonic space and explicit order parameter treatment allows extension to spatially-varying scenarios (interfaces, defects, grain boundaries) and to inhomogeneous or confined systems.
- The rigorous connection to simulation data provides a reliable theoretical foundation for interpreting and guiding scattering experiments, colloidal crystallization, and soft-matter engineering.
Speculation and Outlook:
- Future work can generalize the theory to dynamical regimes, disordered/user-defined lattice structures, and fluctuation-driven transitions.
- Incorporating position-dependent (fluctuating) order parameters may allow for the full spectrum of phase behaviors, including continuous transitions and critical phenomena, to be addressed within the DFT formalism.
- The approach may serve as a template for extending DFT to non-equilibrium, externally-driven, or quantum molecular systems.
Conclusion
This paper provides a rigorous, unified, and practical framework for ordering, correlation functions, and phase transitions in molecular systems, addressing long-standing deficiencies in older density functionals. By leveraging a self-consistent harmonic decomposition and explicit separation of symmetry-breaking correlations, the formalism enables quantitatively accurate phase diagrams and structural predictions for a wide array of soft and hard matter systems. The methodology’s systematic nature, along with its connections to both analytical and numerical solutions, sets a new benchmark for the first-principles treatment of ordering and transitions in complex molecular systems.

