CrystalBoltz: End-to-End Protein Structure Determination via Experiment-Guided Diffusion for X-Ray Crystallography
Abstract: Generative models trained on public databases of protein structures, most of which have been determined by X-ray crystallography, now provide powerful priors for structure prediction. However, they are not readily conditioned on the measurements from a new crystallographic experiment, limiting their use for X-ray structure determination. In crystallography, the measured structure-factor amplitudes do not by themselves determine an electron density map or atomic structure because the associated phases are unobserved and must be inferred. Structure determination therefore remains an inverse problem in which candidate models must be both structurally plausible and consistent with measured diffraction data, often requiring substantial manual refinement by human experts. Emerging methods aim to incorporate experimental information more directly into predictive and refinement workflows. We present CrystalBoltz, a generative framework that casts crystallographic refinement as Bayesian inference over atomic structures and operates directly on structure-factor amplitudes. CrystalBoltz moves from unguided generation with a pre-trained prior over protein structures to experiment-guided posterior sampling, followed by atomic coordinate and B-factor refinement. Across multiple protein crystallography datasets, CrystalBoltz attains lower coordinate RMSD and lower R-factors than the strongest baselines considered, while reducing runtime by a factor of 33 relative to existing experimentally guided refinement.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper introduces CrystalBoltz, a new computer method that helps scientists figure out the 3D shape of proteins using X-ray crystallography data. It combines a smart generative model (a program trained on many protein shapes) with the actual measurements from an experiment, so the final model is both realistic and truly matches the data.
The big questions the paper asks
- How can we use what computers “know” about typical protein shapes and still make sure the result fits the new X-ray data from a specific experiment?
- Can we fix the “phase problem” in X-ray crystallography (missing wave timing information) by guiding a generative model directly with the measurements we do have?
- Can this be done accurately and much faster than current tools?
How did they do it?
To set the scene, here’s a quick refresher and then the two main steps of CrystalBoltz.
A quick crash course on X-ray crystallography
- Imagine shining X-rays through a crystal made of many copies of the same protein. The X-rays scatter and make a pattern of bright spots.
- Each spot has a “strength” (amplitude) and a “timing” (phase) like a water wave. The experiment measures the strengths but loses the timings. That missing timing is the “phase problem.”
- Without phases, it’s hard to go back from the spots to the electron density map and then to the atomic model. So scientists use tricks and a lot of refinement to solve the structure.
Two important ideas:
- Structure factors: these describe how the X-rays scatter at different angles. Think of them as the recipe that tells you how the pattern of spots is formed.
- R-factors: numbers that measure how well your computed pattern from a trial model matches the measured pattern. Lower is better.
The idea of CrystalBoltz
CrystalBoltz mixes two kinds of knowledge:
- A strong “prior” (what proteins usually look like) learned by a generative model called Boltz-2.
- The actual experiment’s data (the measured spot strengths, called structure-factor amplitudes).
It treats structure solving like this: “Find protein shapes that both look like real proteins and match the new data well.” In statistics terms, that’s sampling from a posterior distribution.
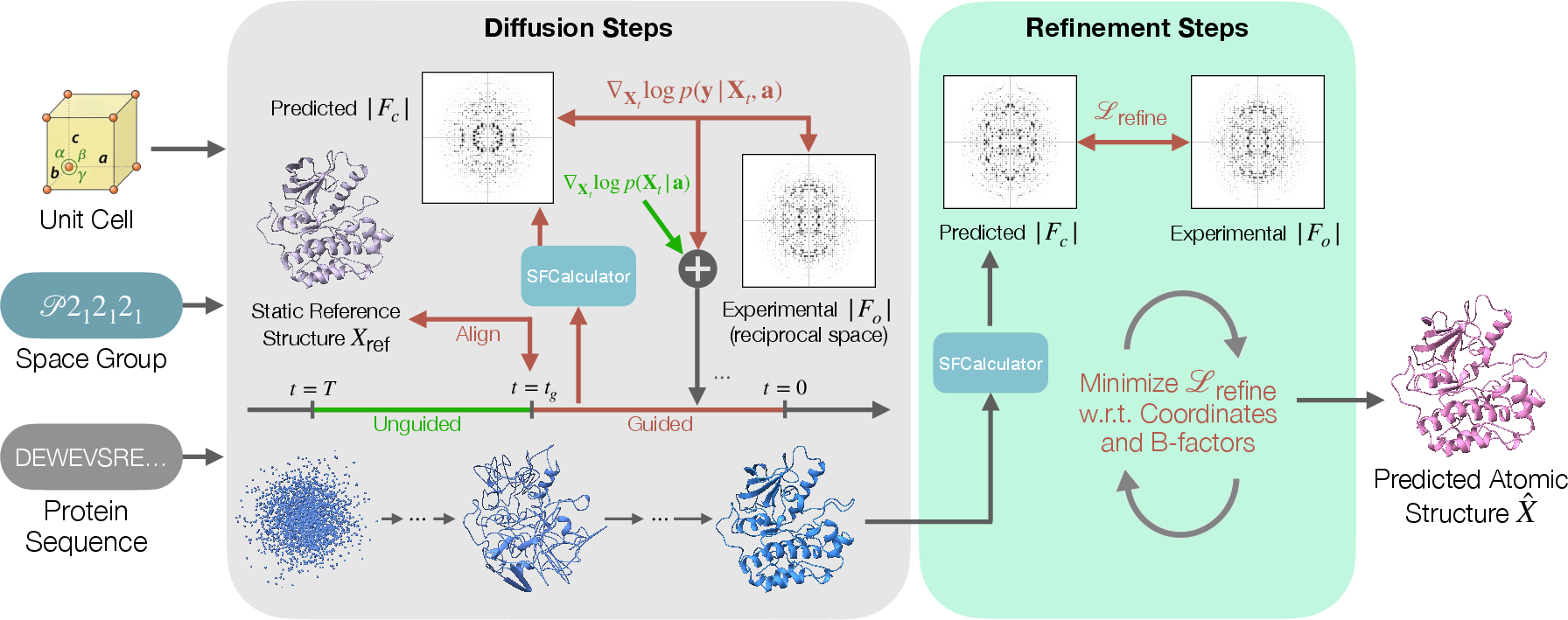
Step 1: Guided diffusion (building a good global shape)
- Diffusion model: Picture starting with a blurry guess and unblurring it step by step. At each step, a neural network nudges the model to look more like a real protein. That’s how modern generative models make 3D protein structures.
- Guidance with data: Once a rough fold (overall shape) appears, CrystalBoltz turns on guidance from the experiment. It compares how well the current guess’s calculated spot strengths match the measured ones and gently pushes the model in directions that improve the match.
- Why start guidance mid-way? Before a shape forms, the model is basically noise, and the data would pull it in misleading ways. Waiting until a coarse shape exists lets the data “fine-tune” rather than derail the process.
A practical detail:
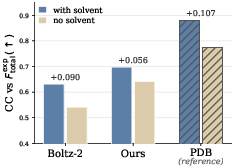
- Bulk solvent: Protein crystals also contain water and other “solvent.” That solvent affects the X-ray pattern. CrystalBoltz includes a differentiable way (meaning the computer can learn from tiny changes) to account for the solvent signal, so the comparisons to data are more realistic.
Step 2: Fine-tuning (refinement of details)
After the guided diffusion makes a strong global model, CrystalBoltz runs a short refinement:
- It adjusts atom positions slightly.
- It optimizes B-factors, which describe how much each atom “wiggles” due to motion or disorder. Think of B-factors as blur settings for each atom: higher means more wiggle/blur.
- This step aims to directly improve the crystallography scores (like R-factors) and fix local details, such as side-chain orientations.
What did they find?
Here are the key results across six real protein datasets:
- Better accuracy vs. other methods:
- Lower RMSD: RMSD measures how far your atoms are from the reference structure (lower is better). CrystalBoltz typically had the lowest RMSD, meaning its models matched the known structures more closely.
- Lower R-factors: Both R_work and R_free (a cross-check on held-out data) were generally lowest with CrystalBoltz, meaning the models agreed better with the experimental measurements without overfitting.
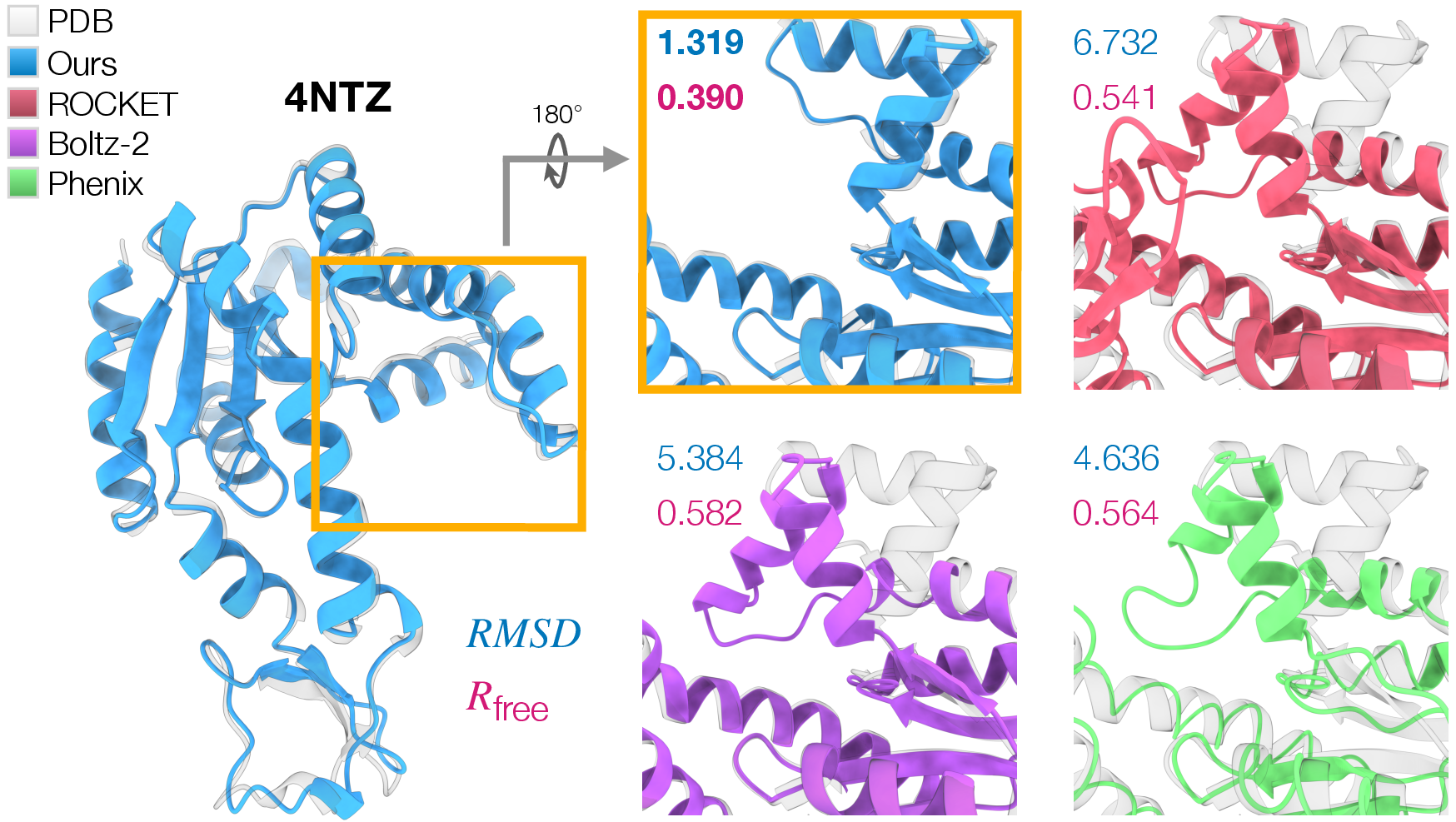
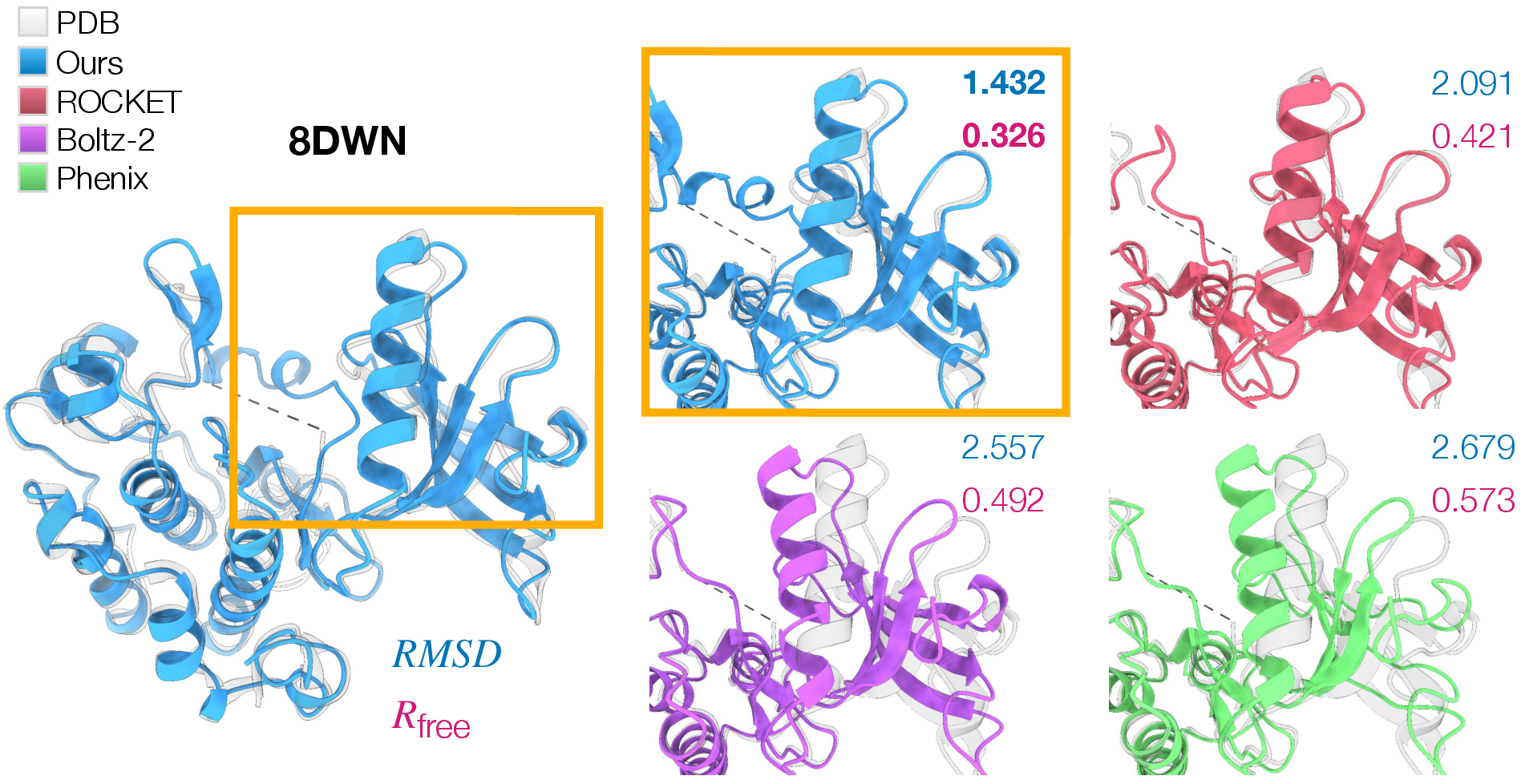
- Especially strong on tough cases:
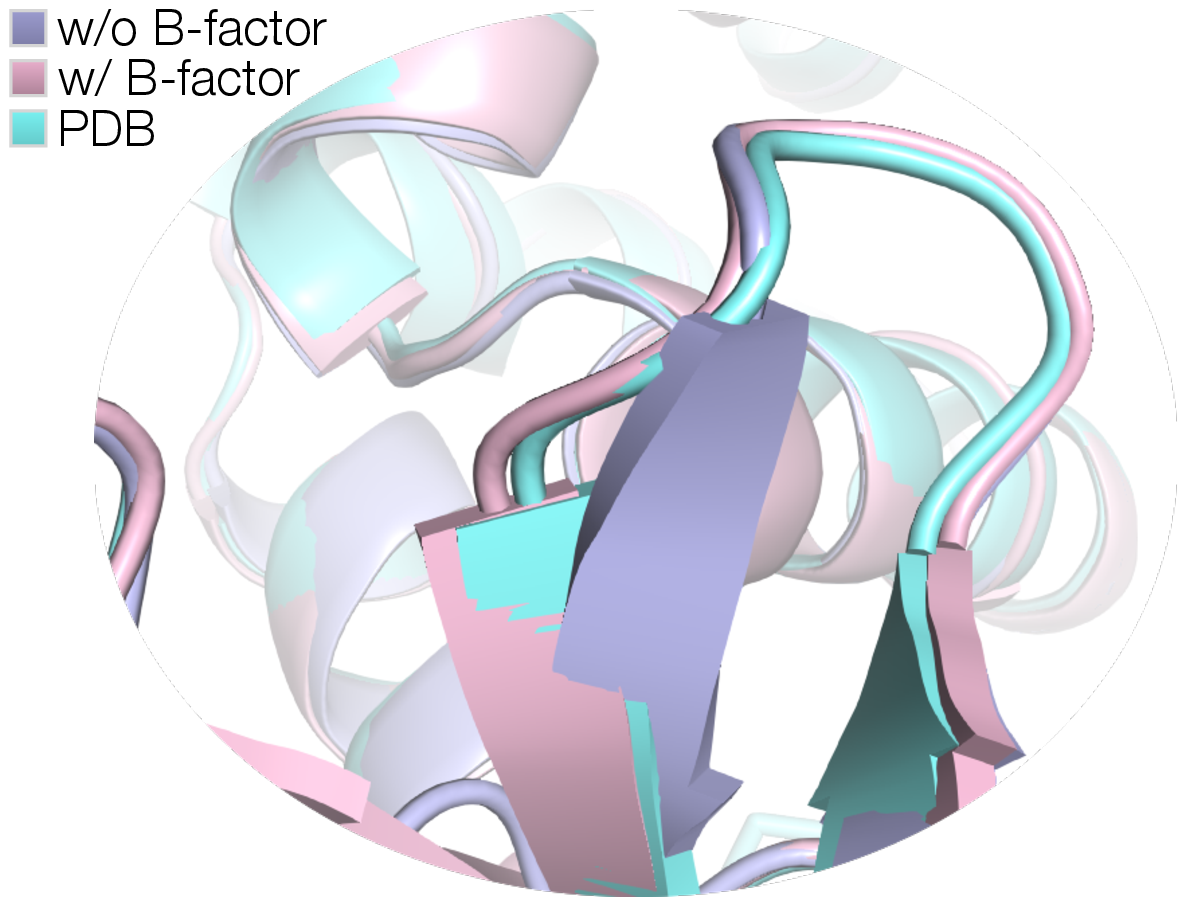
- For proteins where the prior guess was far from the truth (big shape changes needed), CrystalBoltz sharply improved both the 3D match and the fit to the data. In two challenging examples (8DWN and 4NTZ), it cut RMSD by large margins and reduced R_free noticeably.
- Much faster:
- Compared to a strong data-guided baseline method (ROCKET), CrystalBoltz was about 33 times faster on the same kind of GPU (around 11 minutes vs. about 6 hours per target). That speed can make a big difference when data is being collected and quick feedback is valuable.
Why these results matter:
- RMSD: Think “how close is my final 3D model to the known correct shape?”
- R_work and R_free: Think “does my model really match what the experiment saw, even on data I didn’t use to fit it?” Lower is better and reduces the chance of fooling yourself.
Why it matters
- Faster, more reliable structure solving: CrystalBoltz reduces the manual effort and time needed to go from X-ray data to a trustworthy protein model. This can speed up research on how proteins work and how to design drugs.
- Strong combo of knowledge and data: It shows that blending a powerful learned prior (what proteins usually look like) with real measurements (what this specific protein looks like in the lab) gives better and quicker results than either alone.
- Broad potential: The same idea—guiding a generative model with experimental data—could extend to multi-chain protein complexes, other lab techniques (like cryo-EM), and future, even better guidance algorithms.
- Practical impact: Because it’s fast, scientists could use CrystalBoltz during data collection sessions to quickly test and refine models, making experiments more productive.
In short, CrystalBoltz is like having a smart guesser that already knows a lot about proteins, then letting the experiment steer that guesser to the exact answer—accurately and quickly.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a single, concrete list of what remains missing, uncertain, or unexplored in the paper, framed to guide future work.
- Multi-chain and multi-copy assemblies are not evaluated; the method is limited to single-chain asymmetric units. How to place and refine multiple chains and handle non-crystallographic symmetry (NCS) within the guided diffusion framework remains open.
- The approach relies on aligning denoiser predictions to the crystal frame using a “static reference structure,” creating a dependency that is unrealistic for de novo solves. How to perform fully de novo rigid-body placement (orientation and translation) without a reference—e.g., via differentiable Patterson/likelihood searches integrated into guidance—remains unresolved.
- The pipeline assumes known and correct unit cell and space group. In practice, space-group ambiguities and reindexing issues occur; integrating space-group selection/reindexing into posterior-guided sampling is an open problem.
- Guidance acts only on observed amplitudes |F|; the method does not leverage partial phase information (e.g., from SAD/MAD or molecular replacement). Incorporating phase priors/constraints into the likelihood and evaluating their impact on convergence is unexplored.
- The differentiable forward model omits several standard crystallographic effects: anisotropic overall scaling, twinning, anisotropy, radiation damage, and absorption corrections. Extending the forward model and testing on crystals with such pathologies is needed.
- Solvent treatment is bulk-only; no discrete solvent (water) modeling is performed. The large R-factor gap to deposited structures likely reflects missing water placement and refinement; integrating differentiable water building and iterative solvent modeling is a key next step.
- Only isotropic B-factors are refined (clamped to [1, 80] Ų). Support for anisotropic ADPs, TLS/group B-motion, and domain-wise anisotropic scaling is absent; their effect on fit and geometry remains unquantified.
- No occupancy refinement is performed (e.g., for alternate conformations, partial-occupancy side chains, or ligands). Introducing occupancy parameters and testing their identifiability under amplitude-only guidance is an open direction.
- Ligands, metals, post-translational modifications, and other heteroatoms are not handled, yet they often dominate R-factors. Incorporating heteroatom parameterization and differentiable ligand placement/refinement is needed.
- The post-guidance refinement optimizes crystallographic fit (R-factor/CC) without explicit stereochemical restraints (bond lengths/angles, Ramachandran, rotamers, clashes). Evaluating and enforcing geometry (e.g., MolProbity metrics) is necessary to avoid unphysical models.
- The likelihood combines a Gaussian error model and a Rice model with empirically chosen weights and step sizes. There is no principled calibration of loss weights, per-resolution-shell weighting, or adaptive step-size control; automatic, data-driven tuning is needed.
- The Rice likelihood depends on σA; details of its estimation and update are not provided. Adaptive estimation of σA by resolution shell and its interaction with the diffusion dynamics remain to be characterized.
- Early guidance damages performance, and a hand-tuned mid-way activation is used. Developing principled, adaptive guidance schedules (e.g., triggered by pLDDT, backbone formation metrics, or |F|-based CC) is an open problem.
- The DPS approximation p(y|Xt) ≈ p(y|E[X0|Xt]) may be suboptimal for highly nonlinear, nonconvex targets like structure factors. Testing alternative posterior-sampling algorithms (e.g., DAPS, score distillation variants, or proximal samplers) within the same framework is warranted.
- Uncertainty quantification is not provided. Producing posterior ensembles with per-atom uncertainties (coordinates/B-factors) consistent with |F|-data and reporting ensemble-based statistics is an unmet need.
- Evaluation is restricted to six targets (164–306 residues) at 1.69–2.20 Å. Robustness across sizes (large complexes), membrane proteins, poorer resolutions (≥2.5–3.5 Å), incomplete data, and challenging space groups is not assessed.
- Real-time, in-beamline operation is suggested but not validated. Demonstrating performance on streaming, partially collected datasets (incremental reflections, evolving scaling) and defining convergence/early-stopping criteria remain open.
- The method reports mean over the “3 best of 20” samples, but selection criteria are not fully specified and may inadvertently use Rfree information. Designing unbiased sample selection and model averaging procedures that respect cross-validation is needed.
- Overfitting control beyond Rfree reporting is unclear. Investigating the trade-off between maximizing Rwork and preserving generalization (Rfree), including regularization via priors/restraints and stopping rules, remains to be done.
- Geometry-quality metrics (e.g., MolProbity, clashscore, rotamer outliers) are not reported. Quantifying geometry and developing mechanisms to maintain stereochemical quality during guidance/refinement is necessary.
- The contribution of individual missing model components to the R-factor gap (e.g., waters, ligands, anisotropic scaling, ADPs, TLS) is not quantified. A systematic ablation/attribution study would identify the highest-impact additions.
- Only Boltz-2 is used as a prior. Comparing different diffusion priors (e.g., AlphaFold 3, RF3, Protenix) and analyzing how prior strength/calibration affects guidance efficacy is an open question.
- Handling sequence mismatches, tags, truncations, and register errors (common in experimental constructs) is not discussed. Strategies for sequence correction and register shifts under |F|-guided diffusion need investigation.
- Robustness to outliers and negative intensities is not addressed. Incorporating intensity-based targets, robust reflection weighting, and outlier handling could improve stability.
- Scaling and memory footprint for very large asymmetric units (multiple chains/copies) are not characterized. Profiling and optimizing the differentiable SF computations and backprop through the denoiser for large systems is needed.
Practical Applications
Immediate Applications
The following are actionable use cases that can be deployed now, leveraging the paper’s experimentally guided diffusion framework (CrystalBoltz) and its demonstrated runtime and accuracy improvements on single-chain protein crystallography targets.
- End-to-end refinement for single-chain crystallographic structures
- Sectors: healthcare/biotech (structural biology), pharmaceuticals, academia (structural biology labs), synchrotron facilities
- Tools/workflows:
- Add CrystalBoltz as a “DPS-guided refinement” step between molecular replacement and final refinement in Phenix/CCP4 pipelines
- Containerized CLI or GUI plugin for Phenix/CCP4; Slurm/HPC and cloud-ready images for beamline compute nodes
- Benefits: 33× faster than ROCKET in study setup; improved R_free and RMSD with less manual refinement; feasible during beamtime
- Assumptions/dependencies: availability of processed amplitudes (|Fo|), unit cell and space group; single-chain in the asymmetric unit; GPU access; differentiable forward model (SFCalculator) installed; validation needed for ligands and unusual chemistries
- Near real-time beamline feedback for data collection
- Sectors: synchrotron/beamline operations, robotics (automated sample changers), academia/industry users
- Tools/workflows:
- “Beamline-in-the-loop” refinement: run CrystalBoltz after each data-collection segment to inform strategy (dose, rotation range, crystal swapping)
- Integration with LIMS and beamline control (e.g., ISPyB, MxCuBE, Bluesky) to auto-run refinement and display live R_free trends
- Benefits: faster triage of crystals; early detection of misfolds or conformational heterogeneity; efficient beamtime use
- Assumptions/dependencies: stable data-reduction pipeline (e.g., DIALS/xia2) during collection; enough reflections for meaningful guidance; network and compute resources on-site
- Rescue of difficult structure determinations with large conformational differences
- Sectors: pharma/biotech (challenging targets), academia (novel folds), fragment screening campaigns
- Tools/workflows:
- Use CrystalBoltz-guided sampling when AlphaFold- or MR-based starting models fail to agree with data; sample posterior conformations consistent with |Fo|
- Incorporate mid-way guidance scheduling and short post-hoc refinement to escape local minima
- Benefits: improved success rate on conformational outliers; better R_free without exhaustive manual rebuilding
- Assumptions/dependencies: adequate data quality; correct indexing/merging; method validated in paper for 1.7–2.2 Å resolution and single-chain proteins
- Automated quality control and model-bias checks
- Sectors: software (validation tools), academia/industry (structure validation), PDB deposition workflows
- Tools/workflows:
- Run CrystalBoltz alongside conventional refinement to compare posterior-consistent models vs. prior-biased fits
- Flag cases where generative prior and |Fo|-guided posterior disagree substantially
- Benefits: early identification of model bias; improved confidence before deposition; better use of cross-validation (R_free)
- Assumptions/dependencies: standard cross-validation flags present; acceptance of AI-assisted checks in institutional workflows
- Teaching and method prototyping in structural biology
- Sectors: education (graduate courses), academia (methods development)
- Tools/workflows:
- Classroom labs demonstrating the phase problem and posterior sampling with differentiable forward models
- Swap-in of different likelihood terms (Gaussian vs. Rice) and guidance schedules to illustrate algorithmic effects
- Benefits: hands-on understanding of experiment-guided generative modeling; faster iteration in methods research
- Assumptions/dependencies: accessible GPUs; curated example datasets; permissive licensing for diffusion priors and SFCalculator
- Cloud/HPC service for fast refinement at scale
- Sectors: CROs, pharma/biotech, core facilities
- Tools/workflows:
- Offer CrystalBoltz as a managed service (API + batch submission) integrated with ELNs/LIMS; autoscaling to handle screens
- Benefits: consistent, rapid R_free improvements across many crystals; predictable compute costs
- Assumptions/dependencies: data security/compliance (GxP if needed), cloud/GPU availability, SLA around pre/post-processing pipelines
Long-Term Applications
These opportunities require further research, scaling, or development (e.g., broader validation, multi-chain handling, richer forward models, or integration across modalities).
- Multi-chain, multi-copy, and complex crystallographic assemblies
- Sectors: healthcare/biotech (antibody–antigen complexes, PPIs), academia (large assemblies), structural consortiums
- Tools/products/workflows:
- Extend CrystalBoltz to native multi-chain Boltz-2 predictions; incorporate non-crystallographic symmetry and multi-copy asymmetric units
- Support ligands, metals, engineered residues, and membrane protein environments
- Benefits: broader applicability to real-world targets; reduced manual docking/building for complexes
- Assumptions/dependencies: robust chain/stoichiometry handling; differentiable ligand scattering factors and restraints; enhanced solvent/anisotropy models; extensive benchmarking
- Closed-loop autonomous beamline optimization
- Sectors: synchrotron facilities, robotics/automation, national labs
- Tools/products/workflows:
- “Self-driving beamline” where posterior-consistent structures guide dynamic acquisition (e.g., adapt dose, orientation, detector distance) and sample prioritization
- AI agents optimizing experimental parameters to improve R_free in real time
- Benefits: higher throughput and data quality; minimized radiation damage; better use of scarce beamtime
- Assumptions/dependencies: tight integration with control systems; robust failure handling; facility-level validation and safety policies
- High-throughput fragment and ligand screening with AI-guided refinement
- Sectors: pharmaceuticals/biotech, CROs (XFEL/synchrotron fragment campaigns)
- Tools/products/workflows:
- Posterior sampling plus ligand-aware refinement to rapidly assess bound/unbound states and prioritize hits
- Automated ensemble modeling for partial occupancy and conformational heterogeneity
- Benefits: faster hit triage; improved sensitivity to weak binders; reduced crystallographer burden
- Assumptions/dependencies: differentiable ligand scattering, occupancy refinement, accurate bulk-solvent/anisotropic models; rigorous controls to avoid overfitting
- Cross-modality posterior inference (cryo-EM, time-resolved crystallography, integrative modeling)
- Sectors: structural biology core facilities; software vendors; academia/industry research
- Tools/products/workflows:
- Substitute crystallographic likelihood with differentiable real-space cryo-EM targets; integrate pump-probe/time-resolved |Fo(t)| series
- Joint inference across modalities (X-ray + EM + NMR restraints) using shared diffusion priors
- Benefits: unified, data-consistent structural ensembles; better handling of dynamics and disorder
- Assumptions/dependencies: robust differentiable forward models for each modality; standardized data formats; uncertainty modeling across experiments
- Advanced refinement of physical parameters (beyond isotropic B-factors)
- Sectors: software (refinement suites), academia (methods), pharma/biotech (precision refinement)
- Tools/products/workflows:
- Joint optimization of anisotropic B-factors, TLS parameters, occupancy, twinning/anisotropy corrections, and bulk-solvent models within the posterior framework
- Automatic hyperparameter tuning of guidance schedules and likelihood weights
- Benefits: improved fit without overfitting; better partitioning of coordinate vs. disorder signals
- Assumptions/dependencies: stable, differentiable implementations; high-quality data; cross-validation safeguards
- Enterprise-grade AI refinement platform and ecosystem
- Sectors: software vendors, CROs, pharma IT
- Tools/products/workflows:
- End-to-end platform offering dataset ingestion, posterior sampling, refinement, audit trails, and compliance (21 CFR Part 11-ready)
- APIs for ELN/LIMS, versioned provenance for regulatory submissions and PDB
- Benefits: predictable, auditable pipelines; easier deployment across organizations
- Assumptions/dependencies: licensing/IP for diffusion priors; validation SOPs; long-term maintenance and support
- Policy, standards, and community practices for AI-assisted structure determination
- Sectors: policy/funders (NIH, DOE), repositories (PDB), journals, professional societies
- Tools/products/workflows:
- Best-practice guidelines for AI-guided refinement (e.g., mandatory R_free reporting, provenance of AI steps)
- Benchmark datasets and community challenges for experiment-guided generative methods
- Benefits: transparency, reproducibility, and acceptance in the community; fair comparison across methods
- Assumptions/dependencies: community consensus; repository and journal policy updates; open benchmarks and code availability
- Impact beyond structural biology: accelerated design cycles
- Sectors: energy (enzyme engineering for biofuels/biocatalysis), materials (designed proteins), healthcare (therapeutic design)
- Tools/products/workflows:
- Faster structure turnaround feeding design–build–test–learn loops; integration with protein design and docking platforms
- Benefits: reduced cycle times and costs; better R&D ROI and risk management
- Assumptions/dependencies: generalization to diverse targets and conditions; integration with downstream design tools; sustained compute access
Cross-cutting dependencies to monitor
- Method readiness: current validation is on six single-chain targets at 1.7–2.2 Å; broader benchmarking (multi-chain, ligands, lower resolution, crystallographic pathologies) is required for universal deployment.
- Software stack: availability and maintenance of Boltz-2 (or equivalent diffusion priors), SFCalculator, and GPU resources; compatibility with Phenix/CCP4 and facility IT.
- Risk of overfitting/model bias: strict use of R_free and clear provenance to avoid overstating gains; robust uncertainty estimates.
- Licensing and data governance: IP for pretrained models; security/compliance for proprietary datasets; reproducibility and auditability.
Glossary
- acentric reflections: Reflections without inversion symmetry; in crystallography their structure-factor statistics differ from centric reflections. "By the central limit theorem, structure factors are complex normal for acentric reflections and real normal for centric reflections"
- Adam: A first-order stochastic optimization algorithm used to refine parameters via gradient updates. "run for $T_{\mathrm{refine}$ steps of Adam."
- asymmetric unit: The smallest portion of a crystal structure that, via symmetry operations, generates the entire unit cell. "single chain in the asymmetric unit"
- B-factors: Atomic displacement parameters that model thermal motion or disorder, often refined per atom. "we refine the denoised coordinates and isotropic B-factors against crystallographic objectives."
- Brownian motion: Random noise process driving stochastic differential equations in diffusion models. "where defines the noise schedule, represents Brownian motion, and is the score function"
- bulk solvent: The disordered solvent region in a crystal whose scattering must be modeled alongside the macromolecule. "two scattering sources -- the protein of interest and the bulk solvent in the protein crystal"
- centric reflections: Reflections with inversion symmetry; their structure-factor statistics differ from acentric reflections. "structure factors are complex normal for acentric reflections and real normal for centric reflections"
- correlation coefficient (CC): A Pearson-based metric of linear agreement (often between calculated and experimental amplitudes). "Bulk-solvent term consistently improves CC."
- Cα RMSD: Root mean square deviation computed only on alpha-carbon backbone atoms to assess backbone agreement. "Global RMSD and C RMSD (\AA, lower is better) measure all-atom and backbone agreement against the deposited PDB coordinates."
- Debye–Waller envelope: An exponential attenuation factor in reciprocal space that accounts for atomic displacement via B-factors. "B-factors enter the forward model through the Debye--Waller envelope ."
- Diffusion Posterior Sampling (DPS): A method that augments a pretrained diffusion prior with likelihood gradients to sample from a posterior. "Diffusion Posterior Sampling (DPS) formalizes this idea by augmenting a pretrained diffusion prior with likelihood gradients from a forward model"
- electron density map: A real-space representation of electron density obtained by combining amplitudes with phases in Fourier synthesis. "yielding an electron density map when combined with the observed amplitudes."
- free-flag set: A held-out subset of reflections used to compute cross-validated R_free and prevent overfitting during refinement. "we inherit the deposited free-flag set to define the held-out measurements"
- Gaussian error model: A likelihood assumption that measurement noise is Gaussian, leading to a least-squares objective. "The first is a Gaussian error model, which yields a heteroscedastic least-squares loss"
- heteroscedastic: Having non-constant variance across observations; here, per-reflection uncertainties differ. "which yields a heteroscedastic least-squares loss"
- Miller index: A triplet of integers denoting a set of lattice planes (reciprocal-space index) in crystallography. "spatial frequency , also known as the Miller index"
- molecular replacement: A phasing technique that uses a known or predicted structure as a starting model to determine phases. "molecular replacement using similar structures has been the predominant method due to its efficiency and capability to leverage AlphaFold predictions"
- multiple-sequence alignment (MSA): An alignment of three or more biological sequences used to capture evolutionary constraints for structure prediction. "Subsequent work has altered multiple-sequence alignments or sequence profiles to sample alternative conformations"
- MSA embeddings: Learned vector representations of MSA inputs used within neural structure predictors. "optimizing the MSA embeddings using similar likelihood targets."
- occupancy: The fraction of time (or fraction of the model) an atom is present at a site; used in structure-factor calculation. "with occupancy at the real-space position "
- Pearson correlation: A statistic measuring linear correlation between two variables; used to assess agreement with experimental amplitudes. "re-evaluate the Pearson correlation against the experimental amplitudes"
- pLDDT: Predicted Local Distance Difference Test score estimating per-residue confidence from AlphaFold-like models. "They are initialized from Boltz-2 pLDDT scores using the conversion of~\cite{baek2021}"
- Rice distribution: The probability distribution of the magnitude of a (complex) Gaussian random variable; used for amplitude likelihoods with unknown phases. "The second is the Rice distribution, a more sophisticated target in macromolecular crystallography."
- R-factor: A normalized residual measuring disagreement between observed and calculated amplitudes; lower is better. "The R-factor is reported as on the working reflections and on a held-out reflection set"
- R_free: The cross-validated R-factor computed on a held-out set of reflections to assess overfitting. "with serving as the standard cross-validated metric of model fit."
- R_work: The R-factor computed on the working (fitted) set of reflections during refinement. "The R-factor is reported as on the working reflections and on a held-out reflection set"
- RMSD: Root mean square deviation between coordinates, measuring structural difference from a reference. "Global RMSD and C RMSD (\AA, lower is better) measure all-atom and backbone agreement against the deposited PDB coordinates."
- space group: The symmetry group describing all symmetry operations of a crystal lattice. "comprising the unit cell and space group "
- space-filling mask: A mask defining solvent-occupied regions of the unit cell for bulk-solvent modeling. "is conventionally calculated from a space-filling mask that occupies the remaining volume of the crystal's unit cell not occupied by protein atoms"
- stochastic differential equation (SDE): A differential equation involving a stochastic process; in diffusion models it governs reverse-time sampling. "the reverse stochastic differential equation (SDE) is defined as:"
- structure factor: A complex quantity representing the Fourier component of the crystal’s electron density at a given Miller index. "measuring the structure factors, which refer to the scattering at different spatial frequency , also known as the Miller index"
- structure-factor amplitudes: The magnitudes of structure factors measured experimentally, lacking phase information. "measured structure-factor amplitudes do not include phases"
- Tweedie's formula: A relation that uses the score function to estimate the posterior mean of the clean signal from a noisy observation. "we apply Tweedie's formula and approximate the marginal by a point estimate at the posterior mean"
- unit cell: The fundamental repeating 3D volume of a crystal lattice defined by its cell parameters. "comprising the unit cell and space group "
- variance-preserving diffusion model: A diffusion process parameterization where the data variance remains constant over time, used for generative modeling. "Boltz-2 uses a variance-preserving diffusion model for structure generation, where the reverse stochastic differential equation (SDE) is defined as:"
Collections
Sign up for free to add this paper to one or more collections.

