- The paper demonstrates that fine-tuned DrugFormDTA achieved a Pearson correlation of 0.70, outperforming both classical docking and other ML models.
- The study highlights the importance of rigorous data curation and polyprotein mapping to enhance model reliability and generalizability.
- Benchmarking 15 diverse tools revealed strong per-target performance variability, emphasizing the need for consensus, multi-stage screening pipelines.
Motivation and Context
The increasing risk of viral pandemics, combined with the limitations of vaccine-based interventions—including manufacturing lead times, vaccine hesitancy, and lack of effective vaccines for many pathogens—requires robust, rapid antiviral drug discovery pipelines. Repurposing existing drugs presents an attractive route, but economic disincentives and regulatory barriers have limited industrial engagement, especially for broad-spectrum and combination antivirals. The computational community has responded with an expanding suite of open-source datasets, ML models, and docking tools to accelerate in silico ligand discovery and drug-target affinity (DTA) prediction, focusing particularly on small-molecule antivirals targeting viral proteins.
A critical bottleneck remains: the generalizability and real-world predictive reliability of these computational methods, especially outside “data-rich” classes like kinases. This work presents a systematic benchmarking of 15 open-source DTA prediction tools, including both ML-based models (e.g., Boltz-2, DrugFormDTA, FlowDock) and classical/ML-augmented docking tools (e.g., GNINA, Interformer), evaluated on a curated and cleaned dataset comprising 43,005 viral protein-ligand measurements and a contemporaneous test set of 853 compounds spanning 16 viral targets.
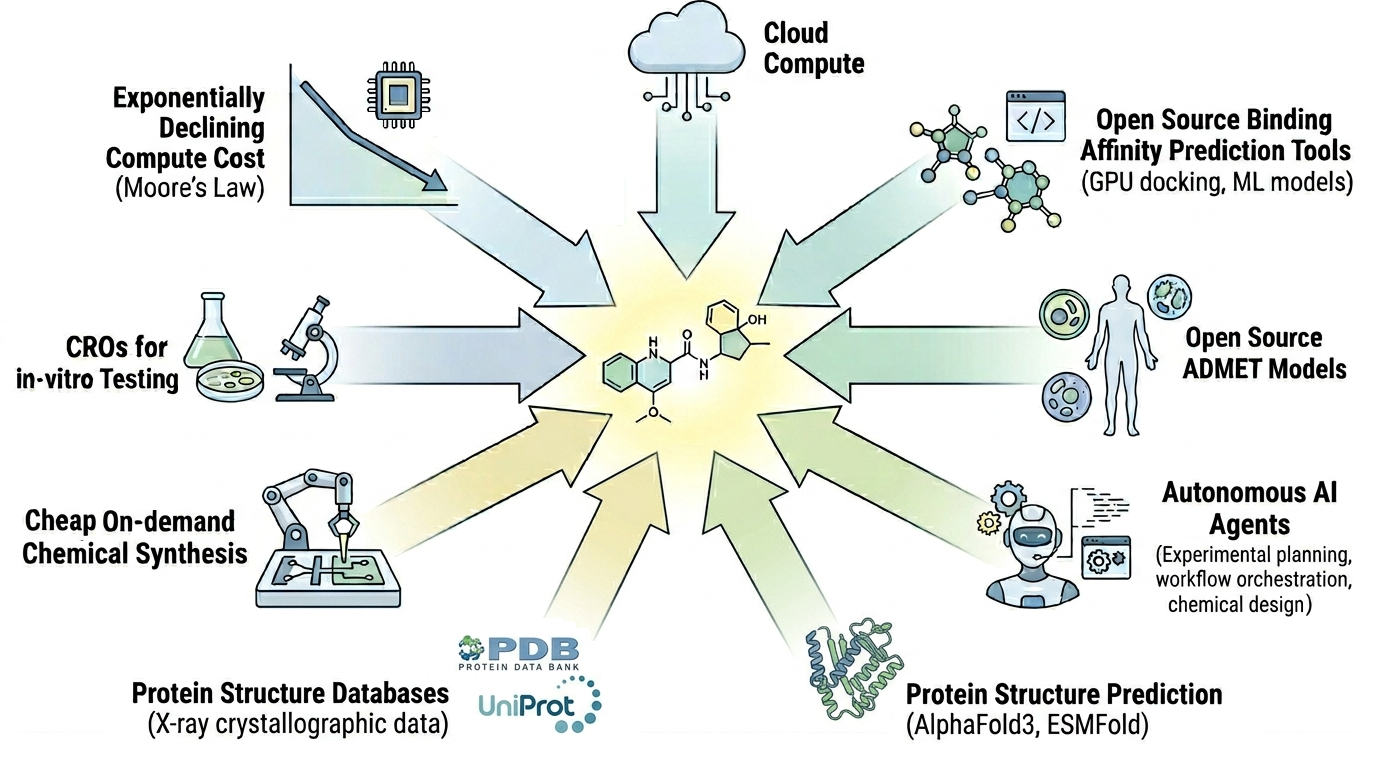
Figure 1: Technologies and trends that are converging to accelerate early stage drug discovery and lower barriers to entry.
Data Resources and Curation
Comprehensive and high-quality datasets are fundamental for both model development and evaluation. The primary sources for protein-ligand affinity included BindingDb, ChEMBL, PubChem, and domain-specific antiviral collections (AntiviralDB, DrugVirus.info, SMACC, Heli-SMACC). Notably, a significant fraction of viral targets in BindingDb are annotated as polyproteins, requiring manual curation and sequence mapping to extract data at the individual functional protein level. Approximately 31% of the BindingDb viral entries required such polyprotein processing before being appropriate for ML model training/testing. Measurement diversity and reliability are also variable: Ki, Kd, and IC50 datasets exhibit high intra-pair value spread, and experimental biases (e.g., assay selection, scaffold clustering) pervade primary data sources.
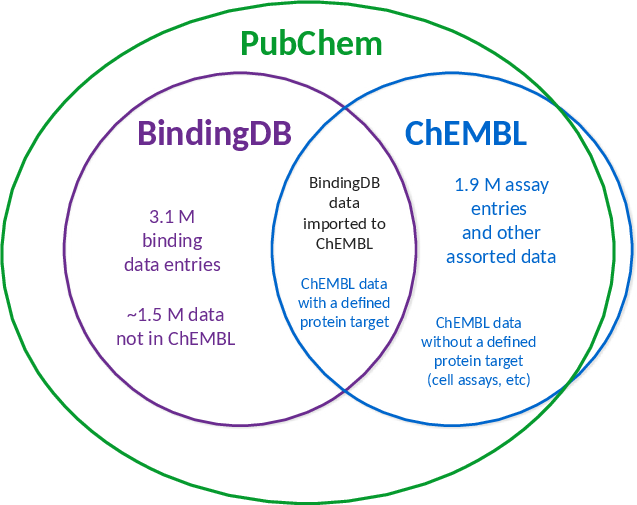
Figure 2: Relation between BindingDb, ChEMBL, and PubChem.
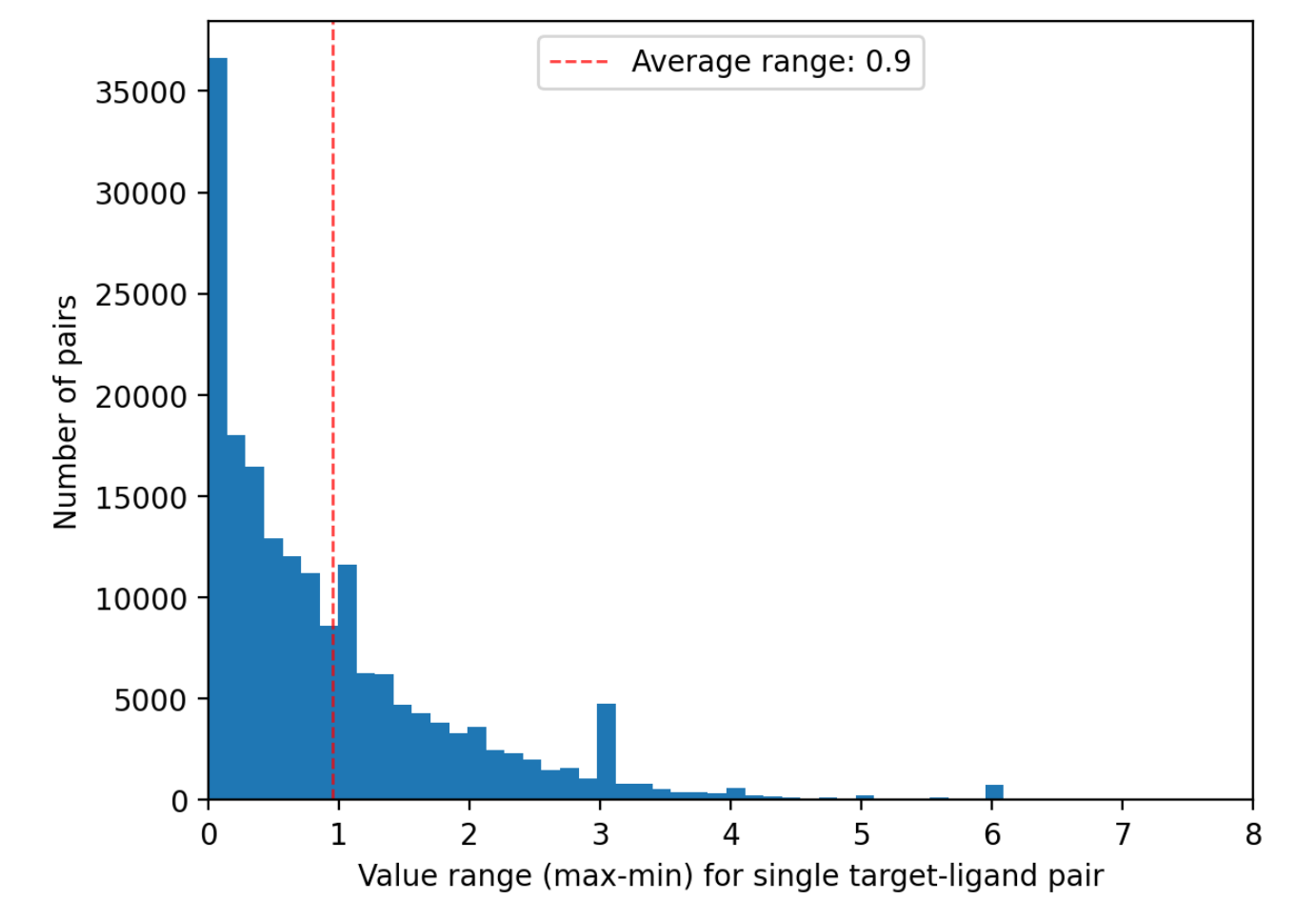
Figure 3: Histogram showing extreme variability in binding affinity measurement ranges across different protein-ligand pairs in BindingDb. The logarithmic x-axis highlights 10x variability per unit.
This work presents a custom, highly curated dataset unifying BindingDb, SMACC, and Heli-SMACC, encompassing 43,005 viral protein-ligand measurement records post-cleaning (including removal of inexact values, unmapped polyproteins, records with missing sequences, and duplicates). These resources underpin the training and testing of both ML and docking approaches.
Survey of Prediction Methodologies
Drug-target affinity prediction can be broadly classified into ML-based, physics/docking-based, and network-based approaches.
ML-Based Methods: Early ML efforts relied on 2D fingerprints and shallow models; the post-2015 era witnessed an explosion of deep learning, graph neural networks, and transformer-based encoders such as Chemformer (for SMILES) and ESM-2 (for protein sequences). Notable contemporary models include DrugFormDTA, Boltz-2, and Protenix/AlphaRank. ML models are highly sensitive to domain shift, clustered protein/ligand scaffolds, and the ratio of active/inactive pairs in training data.
Docking and Physics-Based Scoring: Docking paradigms (e.g., AutoDock, Glide, DOCK, GNINA, Interformer, FlowDock) leverage 3D protein and ligand structures with empirical, force-field, or ML-driven scoring functions to estimate binding affinities. Rigid and flexible docking, blind versus pocket-guided protocols, and consensus scoring approaches all serve distinct roles in virtual screening, with accuracy highly contingent on structural resolution, pocket definition, and the chosen scoring model.
Network Approaches: Graph-based and network-diffusion tools can identify repurposing opportunities from existing drug-disease-protein graphs, but are inherently limited to compounds already present in such networks and are less effective for novel chemical matter or emerging pathogens.
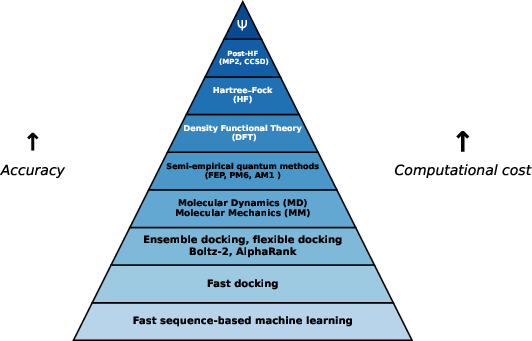
Figure 4: Hierarchy of quantum chemistry methods, augmented with classical physics and docking techniques often used in DTA prediction.
Evaluation and Benchmarking
Test Set Design
The primary benchmark comprises 853 protein-ligand affinity measurements spanning 16 crystal-structure-defined protein targets from 10 viral species, including high-density SARS-CoV-2 and HIV targets as well as less characterized viral proteins (e.g., NS2B-NS3 proteases of Dengue and Zika). Careful mapping of polyprotein-derived targets and the addition of COVID Moonshot and external assay results ensure both depth and out-of-distribution diversity.
Methods Surveyed
Fifteen open-source tools were assessed, encompassing sequence-based ML models (DrugFormDTA, fine-tuned DrugFormDTA, Boltz-2), 3D structure-based ML-docking hybrids (FlowDock, Interformer, GatorAffinity, DrugCLIP, Uni-Mol+GNINA), classical docking (AutoDock-GPU, Vina-GPU, Uni-Dock), and consensus variants. Computational efficiency, input requirements (sequence vs. structure), and pocket targeting (blind vs. focused) were tracked alongside predictive metrics.
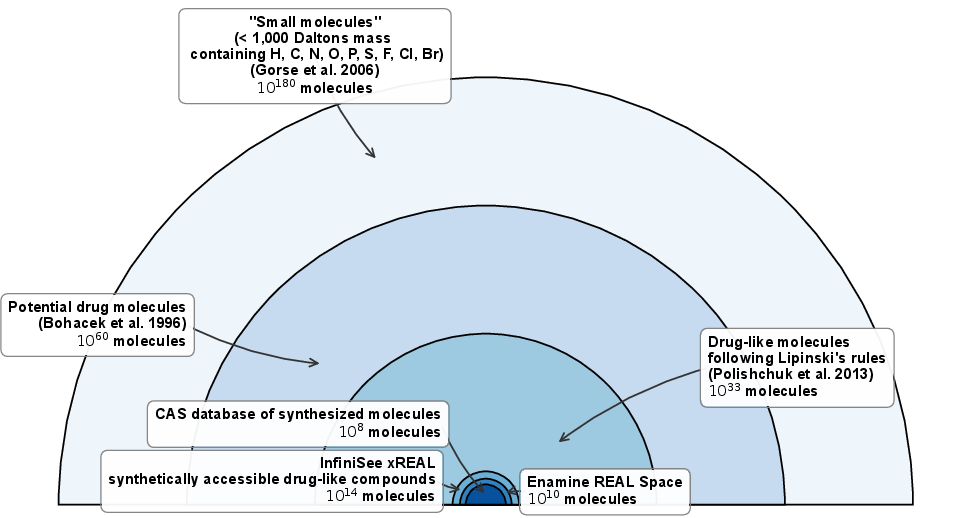
Figure 5: Estimates of chemical space and diversity relevant to small-molecule drug discovery.
Key Results
- Fine-tuned DrugFormDTA exhibited the highest overall test set Pearson correlation (r = 0.70), a marked improvement over its base model (r = 0.50), and robust Spearman ranking (ρ = 0.70). The fine-tuning leveraged the cleaned and split polyprotein dataset constructed in this work.
- Boltz-2 (r = 0.32), GNINA (r = 0.30), and FlowDock (r = 0.30) were the strongest among 3D-structure based methods, with none exceeding DrugFormDTA's sequence-based performance for this data distribution. Importantly, Boltz-2 and GNINA exhibited target-dependent performance volatility, excelling on data-rich proteins (e.g., HIV RT) but lagging on others (e.g., SARS-CoV-2 MPro).
- Rigid docking methods, even with advanced ML scoring, remained inferior to best-in-class ML baseline for this antiviral test distribution (r < 0.25 for all, with occasionally negative r2). Docking algorithms' performance rank strongly depended on target structure.
- Combining DrugFormDTA (fine-tuned), Boltz-2, and GNINA did not significantly exceed fine-tuned DrugFormDTA's stand-alone performance.
- Strong per-target variability was observed (Figure 6): some targets (HIV RT, HCV NS3/NS4A) yielded r>0.6 for multiple methods, while others (SARS-CoV-2 Mpro, Flavivirus NS2B-NS3) saw generally lower predictive agreement across methods.
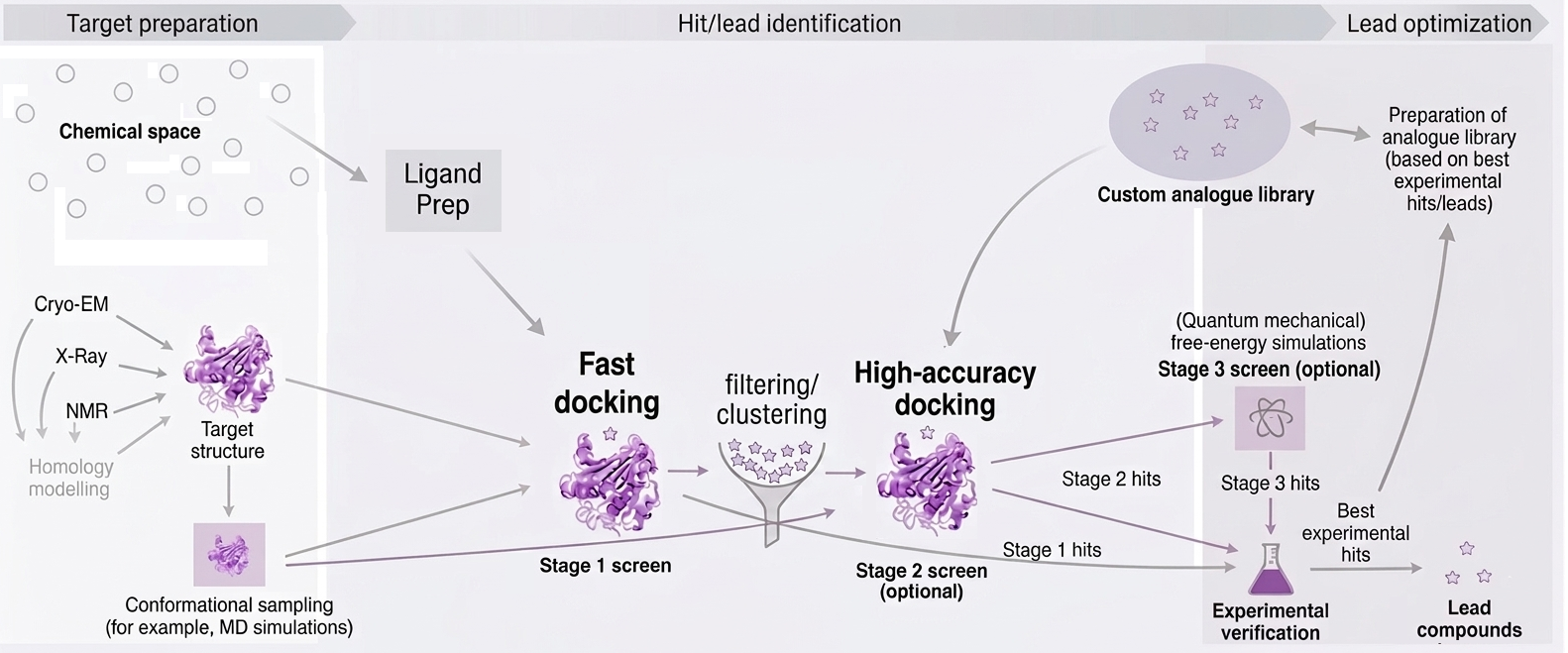
Figure 7: Overview of typical early stage in silico screening workflow, showcasing data integration, method selection, and validation stages.
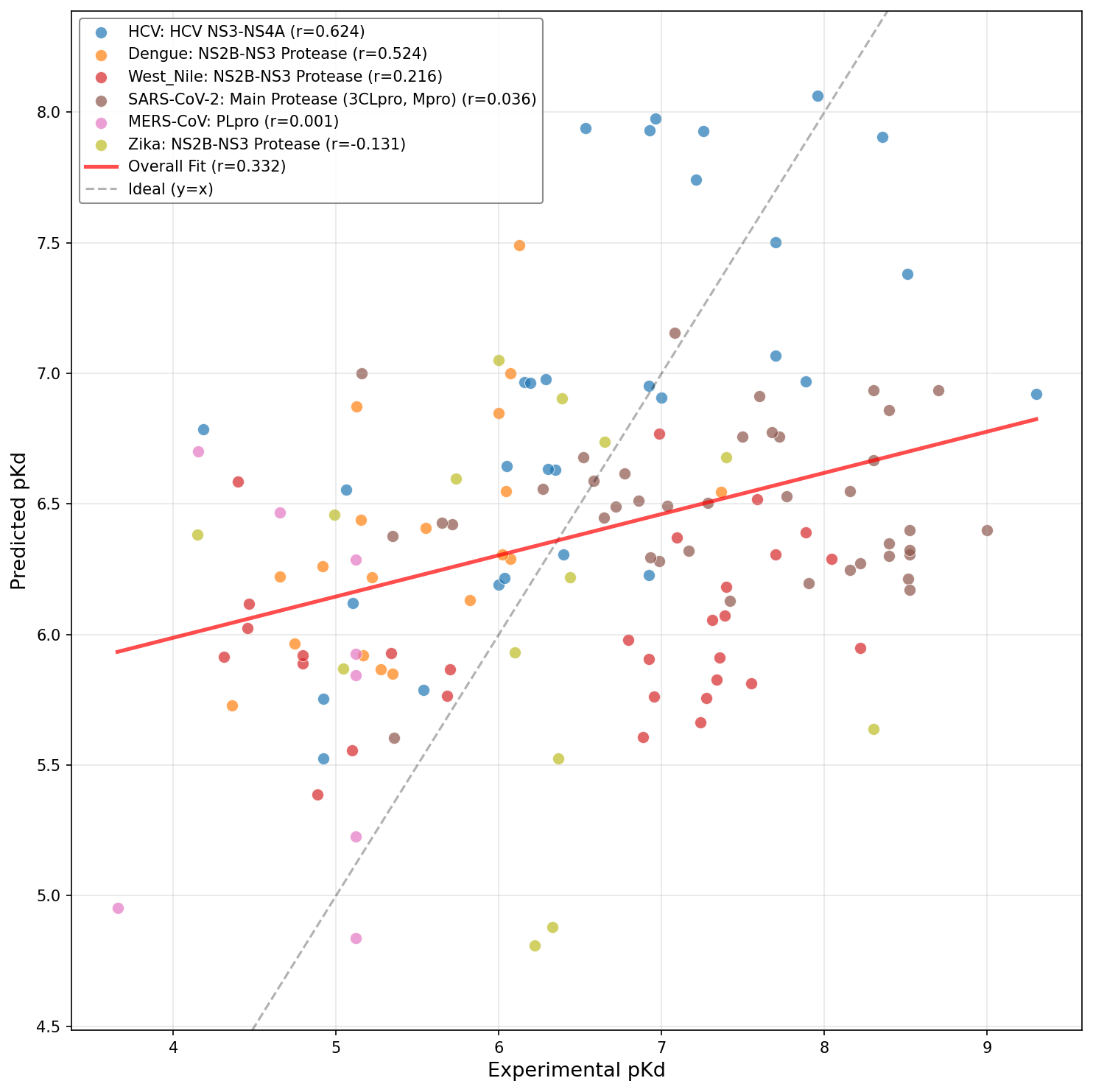
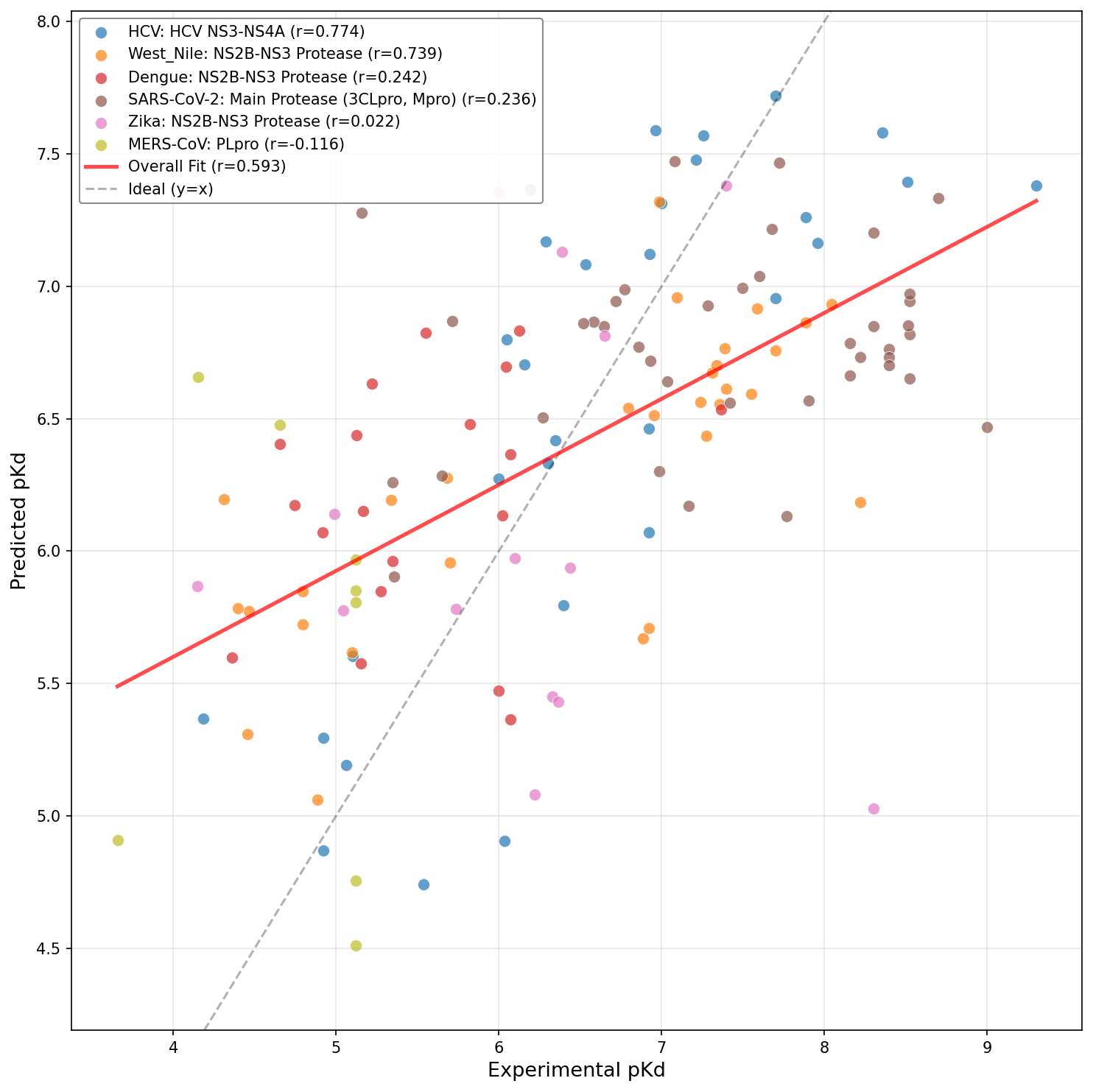
Figure 8: Base DrugFormDTA model.
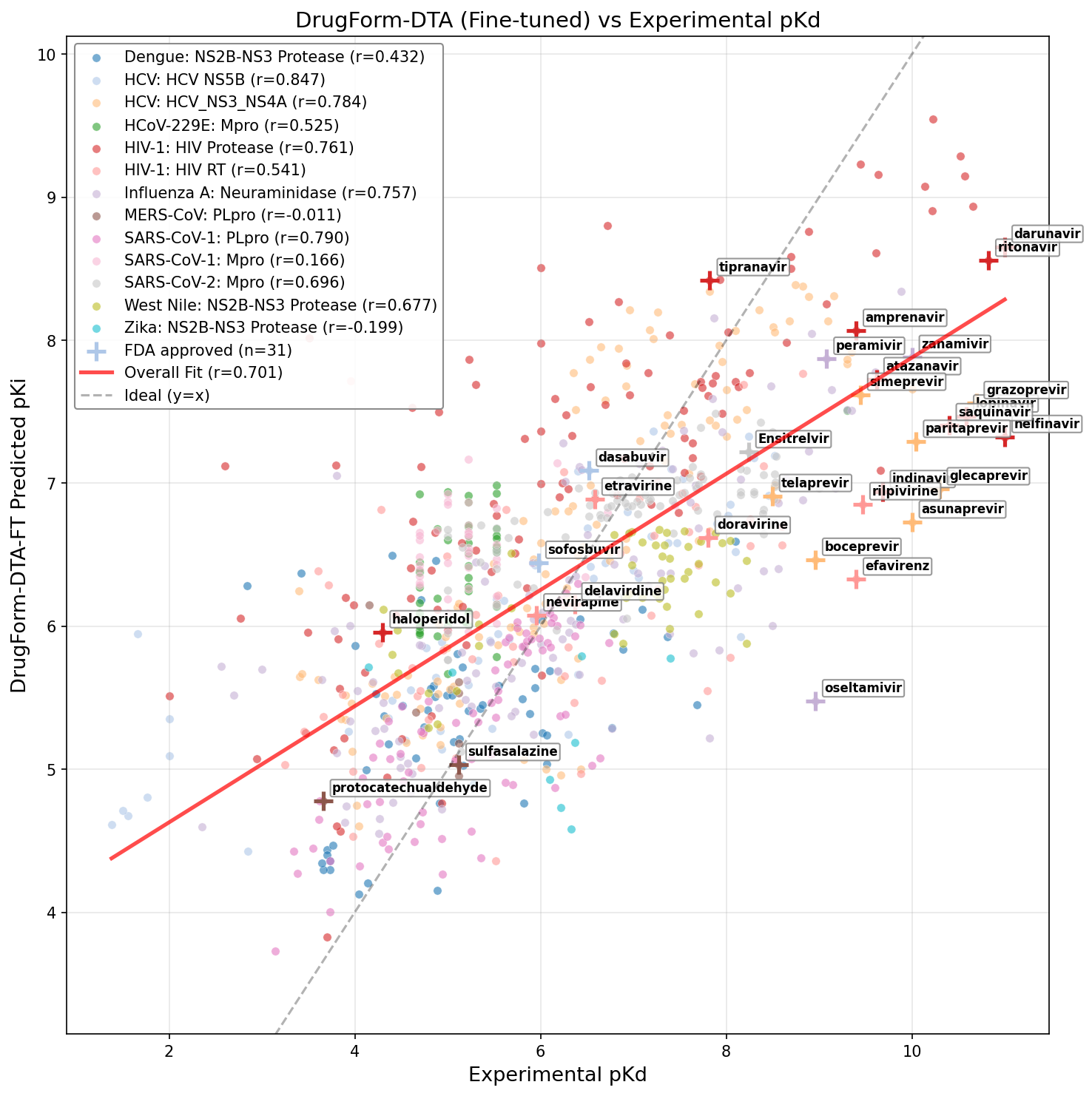
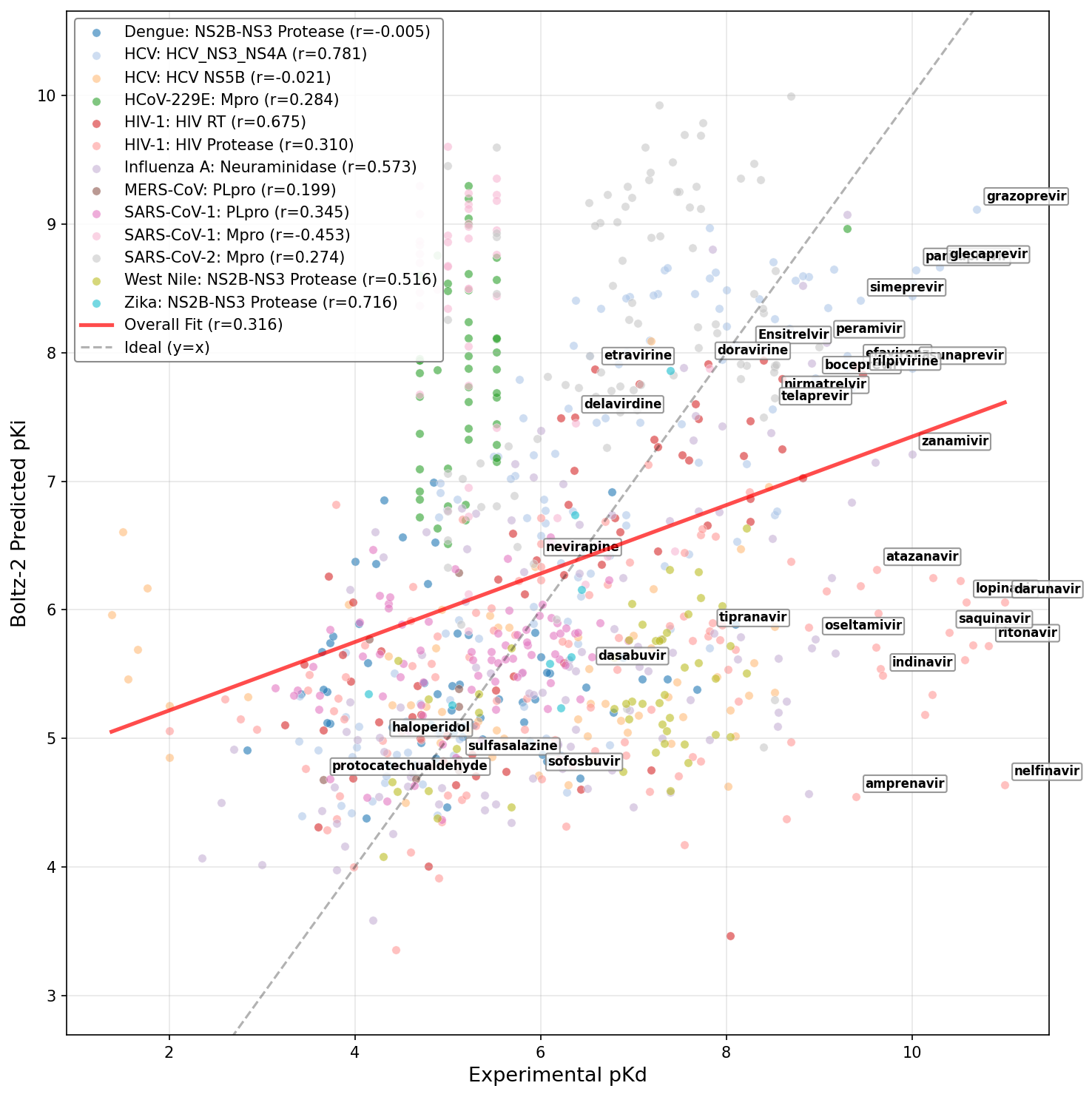
Figure 9: Fine-tuned DrugFormDTA model leveraging curated viral dataset.
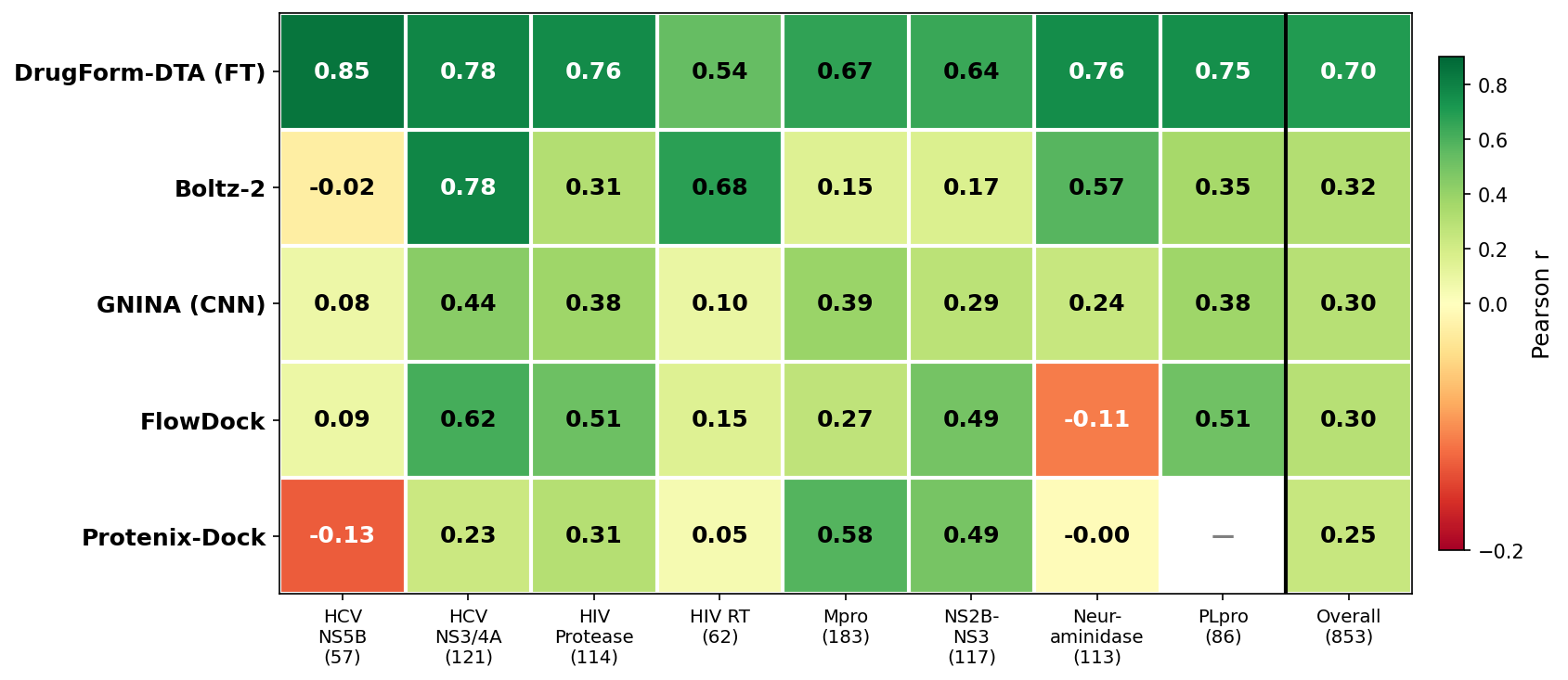
Figure 6: Per-target Pearson r correlation between predicted and experimental pKd/pKi values for each scoring tool. Rightmost column represents overall performance across the benchmark set.
Implications and Future Directions
The results reaffirm several critical points for antiviral drug-discovery researchers:
- Custom data curation and target mapping are essential: Errors and ambiguities in public datasets (notably polyprotein misannotation) can significantly degrade both ML and docking generalization.
- Application-specific fine-tuning on high-quality, diverse data remains essential for robust model performance, especially where the domain diverges from ML model training priors (i.e., away from oncology/kinase targets).
- Hybrid, multi-stage pipelines remain state-of-the-art in hit discovery, consistent with insights from the CACHE challenge series: best results emerge not from any single tool, but from methodologically diverse, multi-filter workflows that combine ML, docking, and expert curation.
- ML models—including “open-source AlphaFold3 analogues” like Boltz-2—still generalize poorly to targets outside their training data distribution, with empirical evidence pointing toward statistical interpolation rather than physics-grounded affinity prediction on novel targets.
- Classical drug-likeness heuristics (e.g., Lipinski, Veber rules) have limited discriminative power for modern antiviral pipelines and should be considered as soft filters rather than strict exclusionary gates.
Practical implications include the rapid identification of repurposable compounds against emerging viral threats—provided that data curation and selection protocols reflect realistic assay conditions, and that computational predictions are validated in multi-stage, consensus-oriented workflows. The benchmarking framework and curated viral affinity resources presented here provide a flexible and extensible foundation for development of open, community-driven antiviral discovery platforms.
Conclusion
A comprehensive, curated benchmarking framework for DTA prediction in antiviral drug discovery has been established. Fine-tuning modern ML models, particularly those using robust molecular and protein sequence encoders, on high-confidence antiviral datasets (with correct target definitions), substantially outperforms both generalist ML and contemporary docking methods in predictive correlation and ranking. Broadly, these insights highlight the necessity of rigorous data cleaning, task-specific adaptation, and ensemble or consensus workflows for effective, scalable in silico antiviral discovery and rapid response to pandemic threats. Continued development and open dissemination of both data resources and benchmarking protocols—as exemplified by this work—remains paramount for translational impact in the field.

