- The paper introduces USB, a simulation-free framework using an unbalanced Schrödinger Bridge approach to model discrete, stochastic birth-death dynamics at single-cell resolution.
- It leverages conditional unbalanced score matching and Poisson-Brownian bridges to achieve scalable and accurate trajectory inference validated on synthetic and real datasets.
- USB demonstrates state-of-the-art performance with near-linear scalability, effectively capturing both diffusive processes and discrete cellular events in high-dimensional settings.
Simulation-Free Reconstruction of Discrete Branching Dynamics: The Unbalanced Schrödinger Bridge Approach
Introduction and Motivation
Single-cell genomics frequently relies on snapshot data due to the destructive nature of sequencing, necessitating inference methods capable of reconstructing underlying cellular dynamics, including both stochasticity and non-conservative mass dynamics such as proliferation and apoptosis. Existing frameworks—most notably, balanced and unbalanced optimal transport (OT), Schrödinger Bridge (SB) formulations, and their simulation-free flow matching extensions—offer only partial solutions, failing to fully integrate stochasticity, unbalance, and the inherent discreteness of cell birth-death events at single-cell resolution.
Addressing these limitations, the Unbalanced Schrödinger Bridge (USB) framework offers a principled, simulation-free approach that models discrete and stochastic cell dynamics at the microscopic level. USB is rooted in the Branching Schrödinger Bridge (BSB) problem, seeking the most likely diffusion and branching process interpolating unnormalized empirical cell state measures across multiple time points. USB introduces an unbalanced score matching training paradigm that enables efficient, scalable modeling and supports biologically realistic simulation of discrete birth-death trajectories.
Theoretical Framework: Branching Schrödinger Bridge and Unbalanced Score Matching
USB builds on the BSB problem, generalizing the classical SB by introducing a reference process of branching Brownian motion (BBM) rather than standard Brownian motion. The BSB seeks the most likely stochastic process (in the form of a BBM) that matches prescribed unbalanced marginal measures μ0,μ1 at initial and terminal time points, minimizing the relative entropy (KL divergence) to the reference process. This setup naturally encodes both stochasticity (via diffusion) and discrete population changes (via birth-death events).
Key distinctions arise between continuous unbalanced OT, which treats mass as a fluid, and the particle-based BBM, where mass is discrete and changes occur via branching or death. The evolution of BBM is described by a Fokker–Planck equation with a source term:
∂tρt=2ν2Δxρt+rρt,
where r parameterizes net proliferation, and the positions follow Brownian motion. For trajectory inference, USB uses conditional Poisson-Brownian bridges for individual Dirac measures—combining stochastic spatial interpolation (Brownian bridge) and linear log-scale interpolation for mass (the continuum limit of a Poisson bridge).
Simulation-free training is achieved by constructing a tractable conditional regression loss—conditional unbalanced score matching (CUSM)—that leverages static semi-couplings between marginal measures (approximated via Wasserstein-Fisher-Rao (WFR) geometry) and closed-form conditional paths, circumventing sample-based simulation of the full SDE/SDE dynamics. The objective jointly regresses the velocity field, growth rate, and diffusion score function, all parameterized by neural networks.
Methodological Implementation
USB’s end-to-end workflow comprises:
- Semi-Coupling Estimation: The static unbalanced coupling between two snapshots is estimated via the static approximation of the RUOT (regularized unbalanced OT) problem, which is itself closely approximated via WFR semi-couplings due to the intractability of the exact BSB/RUOT cost.
- Conditional Path Construction: For each pair in the semi-coupling, the conditional path is a Poisson-Brownian bridge—position evolves according to a Brownian bridge, while mass evolves through a linear-log interpolation, designating the correct birth-death balance.
- Simulation-Free Regression: Network parameters for velocity, growth, and score fields are optimized by minimizing the CUSM objective, which exploits analytic conditional distributions, enabling direct and scalable training in high-dimensional settings.
- Branching and Continuous Inference: For downstream applications, USB supports both continuous (diffusive, population-level) inference via SDE simulation and discrete, single-cell branching simulations reflecting stochastic division and apoptosis events.
Empirical Validation
USB exhibits state-of-the-art performance in trajectory inference across a suite of both synthetic and real single-cell datasets, with comprehensive quantitative and qualitative assessments:


Figure 3: Learned trajectories on the Simulation Gene dataset (ν=0.001) for varying δ.
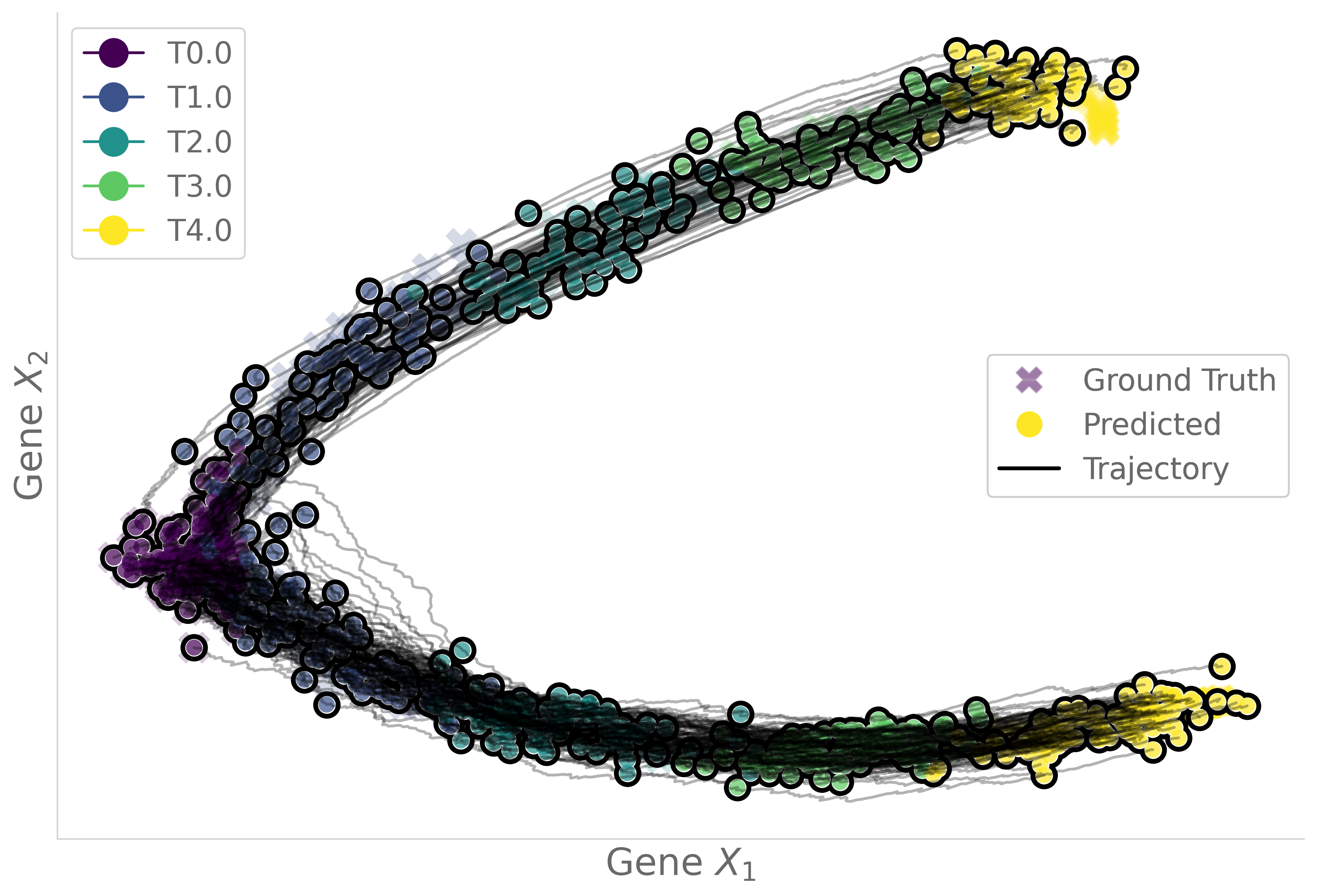
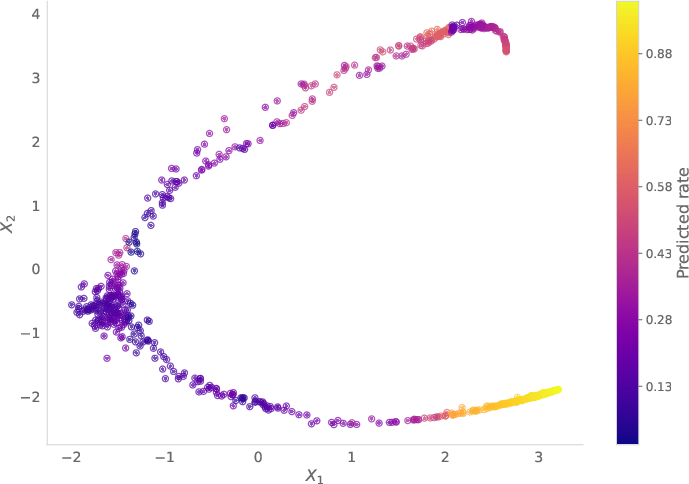
Figure 4: Learned trajectories and growth on the Dyngen dataset (δ=1.7, ν=0.1). Left: trajectories; Right: growth rate.
- Single-Cell Birth-Death Simulation: Through branching inference, USB enables generating explicit, discrete birth-death trajectories at single-cell resolution, visualizing apoptosis, division, and stochastic fate decisions (Figure 5). This capacity is not captured by continuous unbalanced OT or population-based SB extensions.
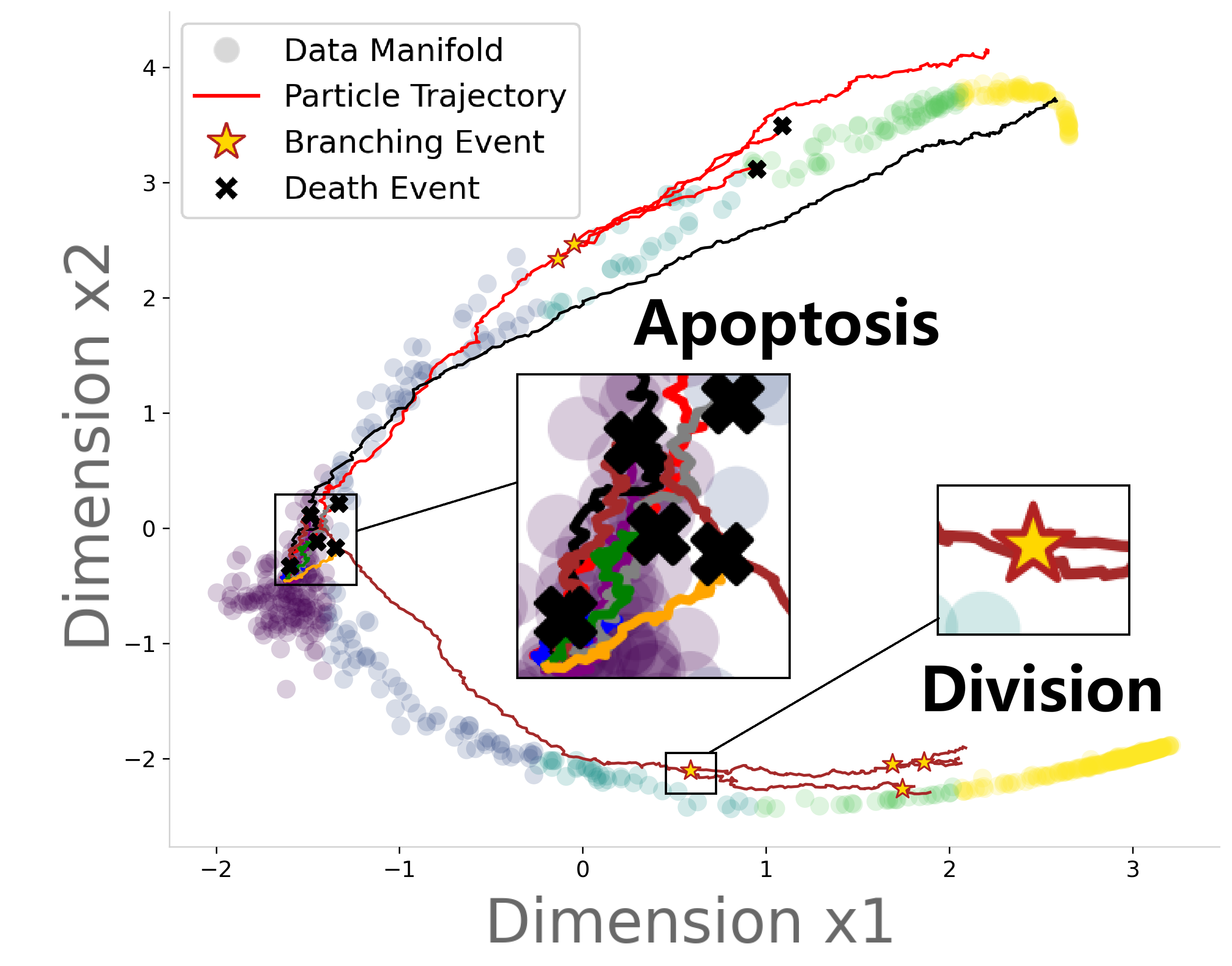
Figure 5: Single-cell resolution birth-death dynamics simulation.
- Scalability: USB exhibits near-linear scaling with both dataset size and feature dimensionality, maintaining competitive or superior runtime versus prior methods (Figure 6).
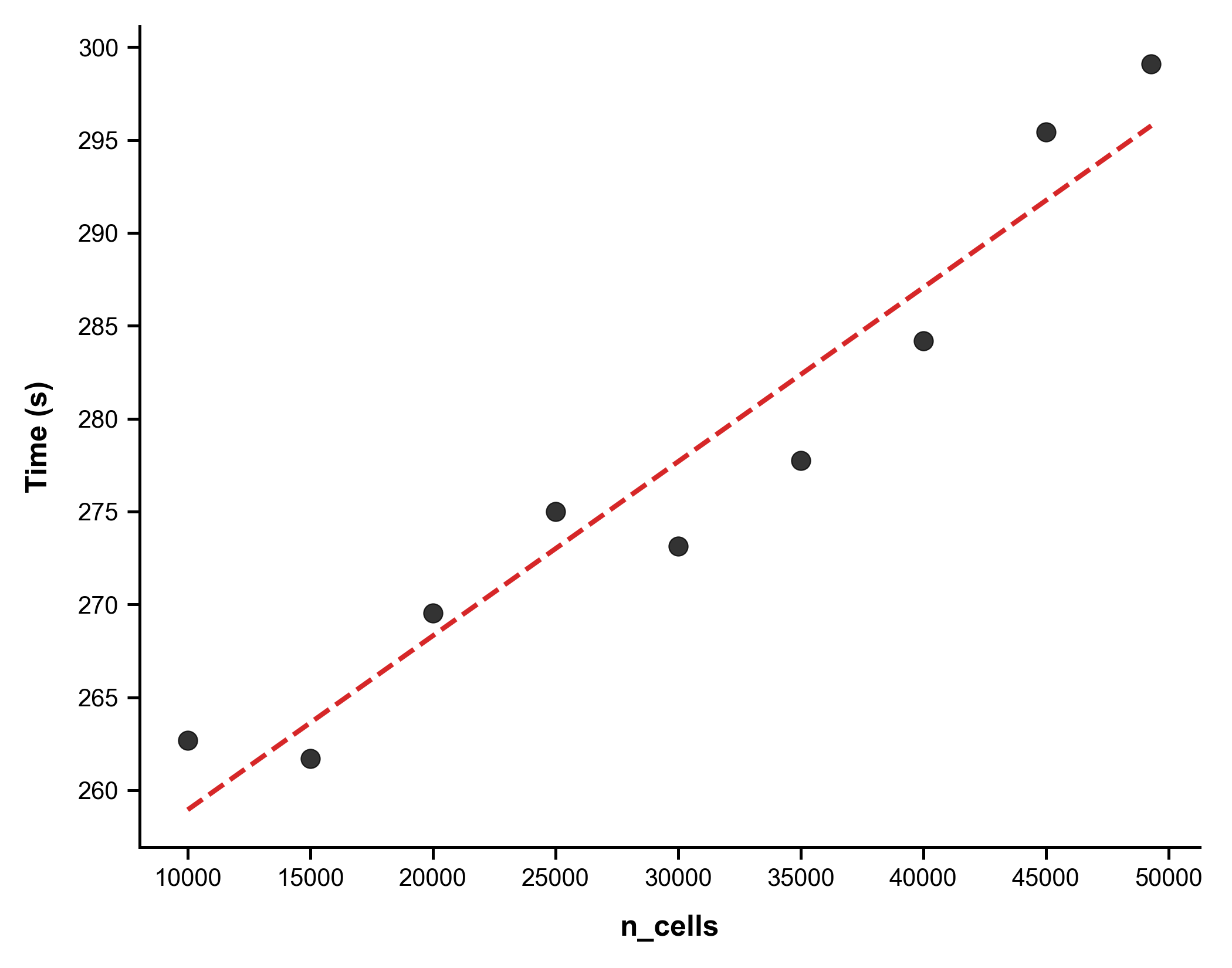
Figure 6: Training time versus cell numbers, demonstrating linear scalability.
Claims, Limitations, and Comparisons
USB demonstrates, with strong empirical evidence, simultaneous modeling of stochasticity, unbalance, and discrete birth-death events, with computational scalability across high-dimensional and large datasets, a combination not achieved in prior work.
Comparative analyses highlight:
- Existing stochastic unbalanced OT and SB solvers (e.g., DeepRUOT) require simulation or expensive iterative procedures, limiting scalability ([DeepRUOT], [UDSB]).
- Simulation-free flow/score matching approaches (e.g., [SF²M], [wfr_fm], [VGFM]) lack one or more key attributes: stochasticity, unbalance, or discrete dynamics.
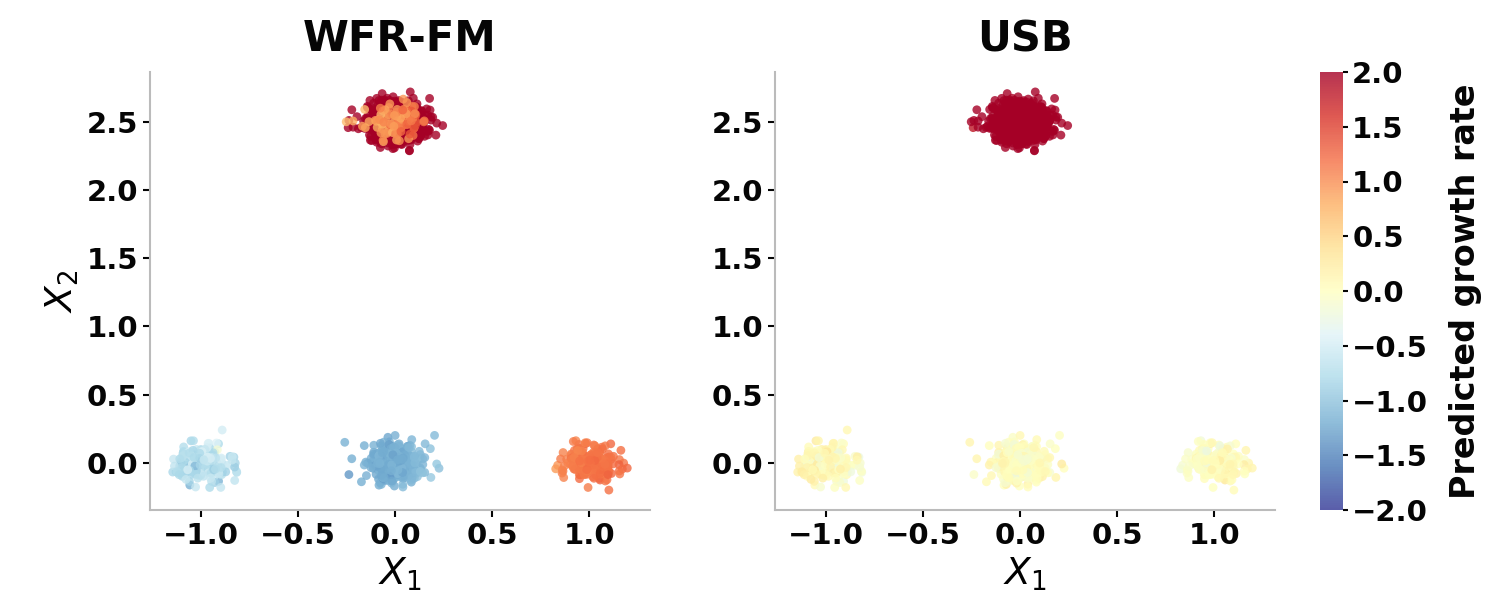
- USB is strictly more expressive, able to reduce to previous models in special cases (e.g., standard SB or WFR-FM as limiting regimes; see Appendix D).
A limitation is the reliance on WFR semi-couplings as an approximation to the BSB/RUOT semi-coupling, justified by analytical intractability of the general Dirac-to-Dirac unbalanced cost (see Appendix: Travelling Diracs). This is a mathematical directions for future exploration.
Implications and Future Directions
Practical implications: USB establishes a new standard for inference of cellular dynamics from purely cross-sectional single-cell data, making it the first simulation-free, scalable, and biologically faithful framework capable of reconstructing—not merely population-level flows, but individual cell fate decisions and stochastic birth-death trajectories.
Theoretical implications: USB formalizes the interface between path-space entropy minimization with respect to branching reference measures and efficient regression-based learning, linking Fokker–Planck approaches, SB methods, and efficient score-based generative frameworks.
Future developments may target: (i) improved analytic approximations or solvers for the static semi-coupling in BSB/RUOT, (ii) extension to more complex cell fate branching structures, (iii) integration with multi-omics modalities, and (iv) broader applications in stochastic unbalanced modeling of discrete systems.
Conclusion
USB fundamentally advances the scope of simulation-free trajectory inference by achieving a rigorous, scalable, and biologically accurate modeling of stochastic unbalanced dynamics, uniquely enabling explicit simulation of discrete birth-death dynamics at single-cell resolution. It broadens the modeling capacity beyond fluidic interpretations to reconstruct genuinely discrete proliferative and apoptotic events, thereby enhancing both theoretical understanding and empirical applicability of computational methods in single-cell dynamic systems.