- The paper demonstrates that extended spin Hamiltonians including biquadratic and Dzyaloshinskii–Moriya interactions are essential for modeling the low-energy excitations of Cr8 and V8 rings.
- It employs non-collinear DFT with DFT+U+V to extract anisotropic exchange parameters, confirming improved experimental susceptibility agreement especially at low temperatures.
- The study highlights that curvature-induced DM effects and competition between FM and AFM couplings fundamentally govern the quantum magnetic behavior of molecular rings.
Magnetic Interactions and Spin Order in Cr8 and V8 Molecular Rings from Non-Collinear ab initio Calculations
Introduction and Motivation
This work presents a comprehensive non-collinear DFT investigation of the magnetic interactions in Cr8 and V8 octanuclear ring-shaped molecular magnets (2604.23565). These molecular systems serve as prototypical quantum magnets for exploring the intricate interplay between geometry, electronic correlation, and spin interactions at the molecular scale. The motivation stems from the recognized inadequacy of collinear spin models, particularly the isotropic Heisenberg Hamiltonian, for fully describing the low-energy excitations in such systems. The authors extend their previous work by employing a non-collinear DFT framework, allowing explicit treatment of arbitrary spin arrangements, and systematically evaluate beyond-Heisenberg couplings, such as biquadratic and antisymmetric (Dzyaloshinskii-Moriya, DM) exchange, as well as their dependence on ring curvature.
Methodology and Model Systems
The Cr8 and V8 molecular rings are constructed from eight transition metal M3+ ions (M = Cr or V), each octahedrally coordinated and connected via bridging fluorine and carboxylate ligands, forming nearly planar octagons with a high degree of local symmetry.
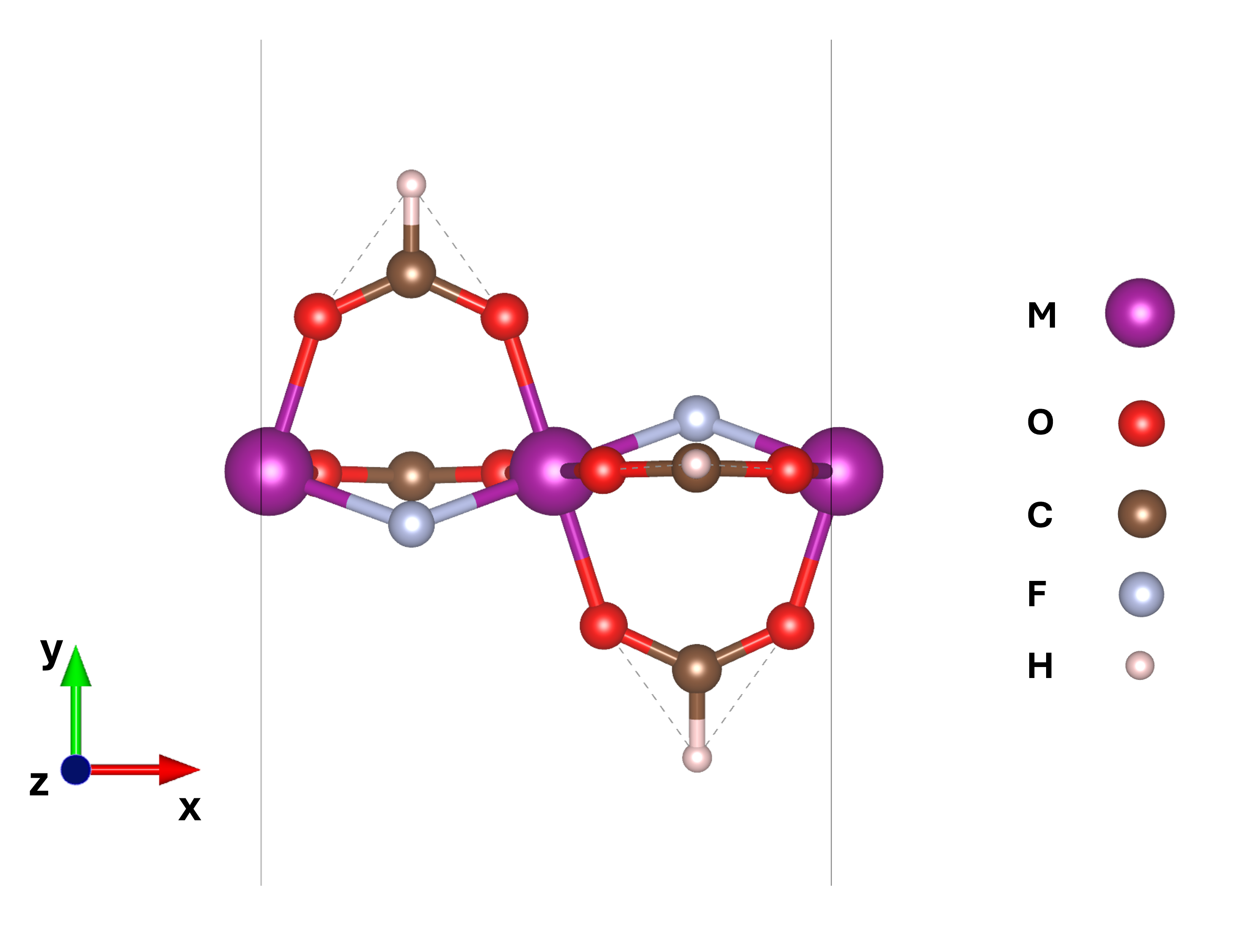
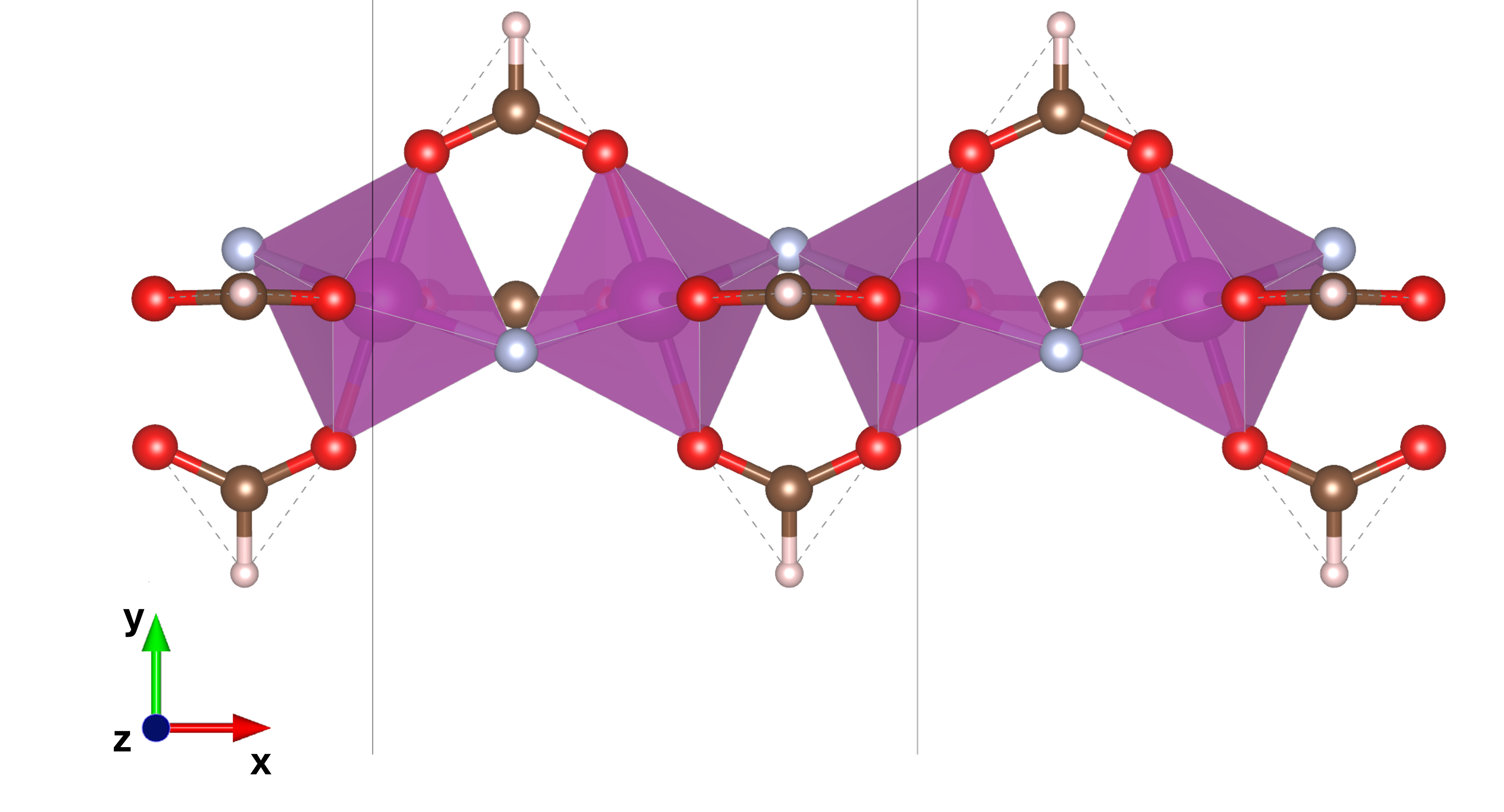
Figure 1: Representation of the M-chain molecular structures, unit-cell and supercell, adopted in this study.
To isolate the effect of structural curvature on magnetic couplings, the authors introduce linear chain analogues with identical local bonding topology. All electronic structure calculations employ plane-wave DFT (as implemented in Quantum Espresso), using both the DFT+U and extended DFT+U+V schemes, with site- and bond-resolved Hubbard parameters extracted via linear-response theory. Non-collinear magnetic states are generated for systematic fitting of spin Hamiltonians against total energies.
Generalized Spin Hamiltonian and Extraction of Couplings
The fundamental advance in this study is the adoption of a spin Hamiltonian of the form:
80
where 81 indexes cylindrical coordinates (82, 83, 84), 85 represent anisotropic exchange, 86 the corresponding biquadratic terms, and 87 the DM vector.
Non-collinear DFT configurations are mapped onto combinations of anti/ferromagnetic and helical spin arrangements, enabling extraction of parameters up to fourth-nearest neighbor and analysis of in-plane versus out-of-plane anisotropy.

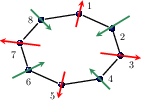
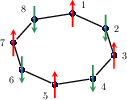
Figure 2: The three spin AFM non-collinear reference configurations.
Principal Results: Exchange, Biquadratic, and DM Interactions
Exchange: Directional Anisotropy and Range
The Cr88 system displays isotropic, antiferromagnetic (AFM) 89 interactions (e.g., 80 meV with DFT+81+82), with negligible further-neighbor couplings. By contrast, V83 exhibits ferromagnetic (FM) 84 (e.g., 85 meV with DFT+86+87) but sizeable antiferromagnetic 88, resulting in strong competition between nearest- and next-nearest-neighbor couplings. Ring curvature ensures cylindrical, rather than Cartesian, anisotropy; exchange is modestly stronger out-of-plane (89) than in-plane.
Biquadratic Couplings
Fitting spin-rotation energetics reveals negligible biquadratic terms for Cr80, but substantial BQ couplings for V81 (e.g., 82 meV, 83 meV in DFT+84). These favor in-plane spin alignment and introduce additional anisotropy, nontrivial in magnitude compared to the leading bilinear terms.
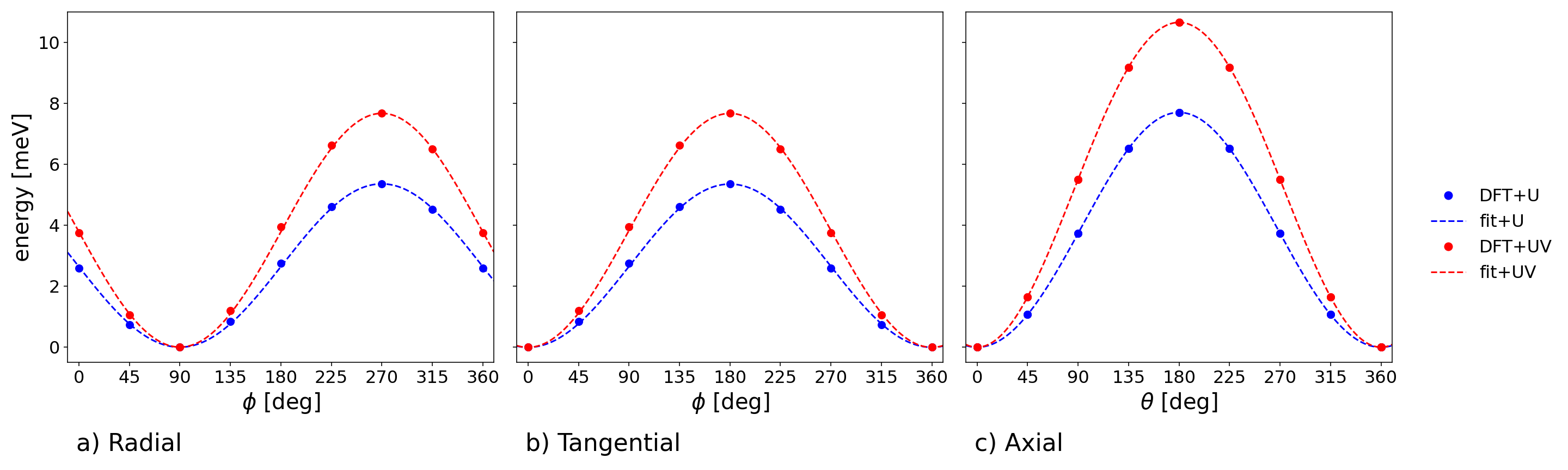
Figure 3: Non-collinear DFT total energy profiles obtained rotating a single spin in Cr85 from the AFM ground state configuration in each direction.
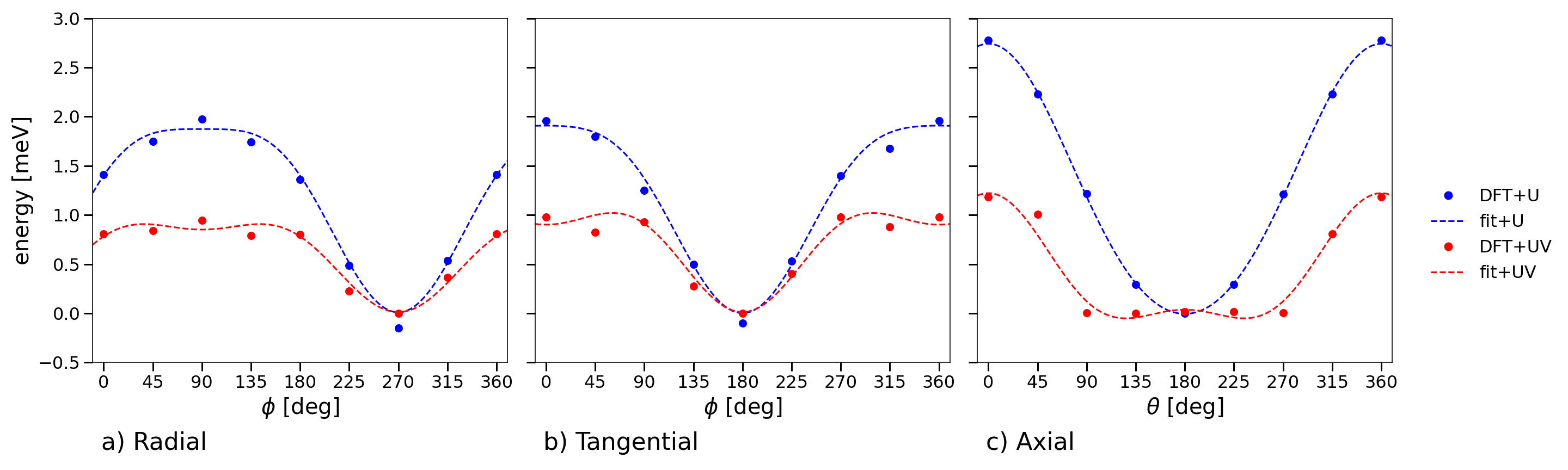
Figure 4: Non-collinear DFT total energy profiles obtained rotating a single spin in V86 from the FM ground state configuration in each direction.
Dzyaloshinskii–Moriya (DM) Interactions
Despite the absence of explicit spin-orbit coupling (SOC) in the calculations, significant DM-like antisymmetric exchange arises, particularly for the 87-component in both rings (88 meV for Cr89 ring, 80 meV for V81 ring with DFT+82+83). These DM terms are attributed in Cr84 primarily to geometric effects (curvature-induced basis transformation), as chain analogues show vanishingly small values. For V85, however, DM couplings are nonzero even in the chain, indicating a possible intrinsic electronic origin unrelated to geometrical curvature.
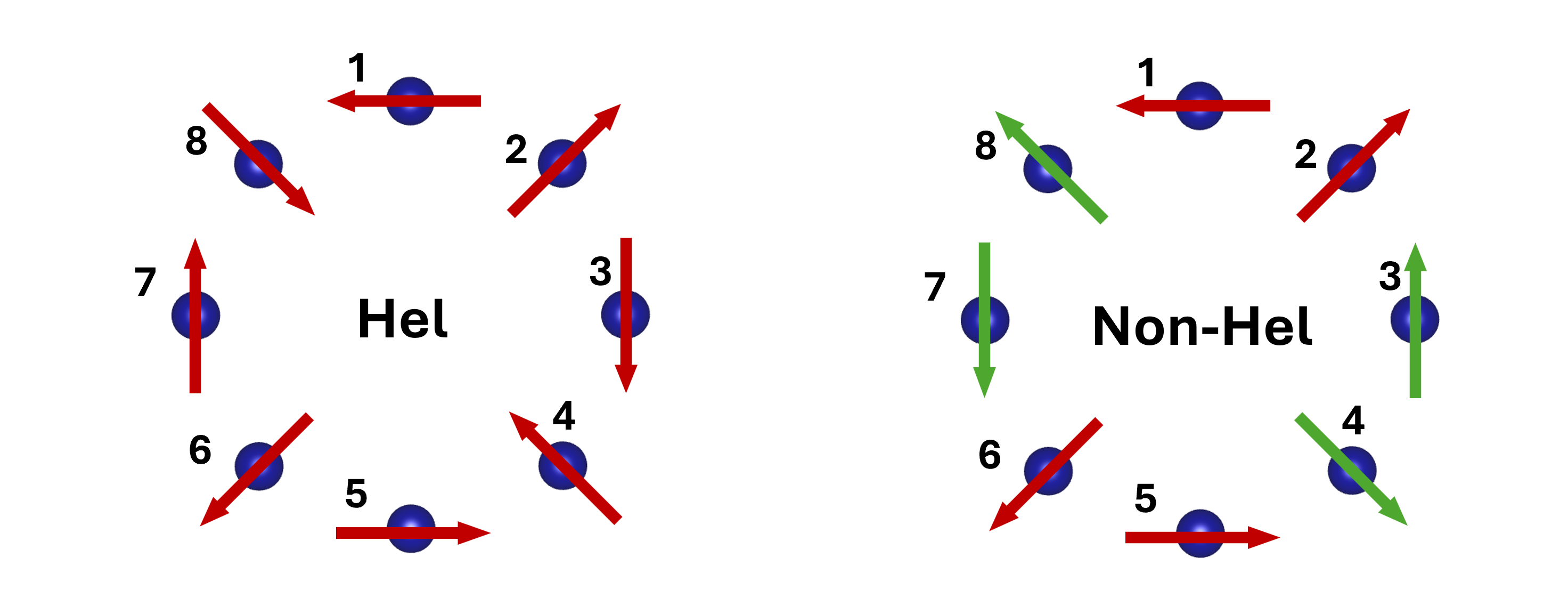
Figure 5: Magnetic spin configurations used to evaluate 86 in Cr87 and V88.
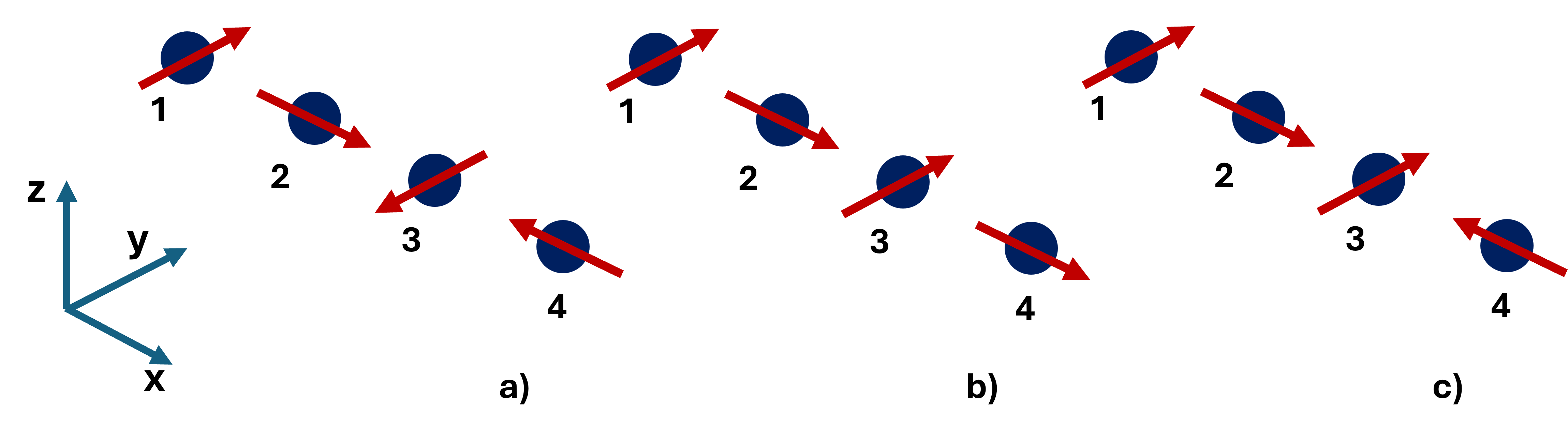
Figure 6: Helical and non-helical spin configurations used to to evaluate the 89 antisymmetric exchange couplings in chain model.
Quantitative Agreement with Experiment and Hamiltonian Validation
Exact diagonalization of the fitted spin models enables direct calculation of the magnetic susceptibilities, which are compared to experimental data for Cr80. The comprehensive Hamiltonian—including extended exchange, biquadratic, and DM terms—matches experimental 81 curves over a broad temperature range, particularly improving low-82 agreement (below 10 K), as shown in the right panel of Figure 7. This endorses the necessity of including geometrically-induced DM and higher-order interactions even in nominally "simple" molecular magnets. DFT+83+84 reproduces experimental trends more accurately than standard DFT+85, confirming the importance of inter-site correlation treatments.
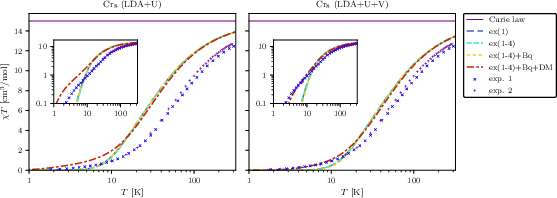
Figure 7: Magnetic susceptibility of Cr86 for different model Hamiltonian truncations alongside experimental data; DM and biquadratic terms are needed for quantitative agreement at low 87.
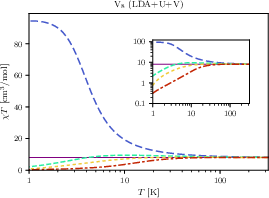
Figure 8: Magnetic susceptibility of V88 for the DFT+89+M3+0 Hamiltonian, highlighting the transition from FM to AFM-like low-M3+1 behavior upon including longer-range interactions.
Strikingly, for VM3+2, the inclusion of antiferromagnetic M3+3 couplings completely inverts the low-M3+4 susceptibility behavior from FM to AFM-like, despite all ab initio predicted interactions being either FM or weakly AFM. This transition, and the associated radical restructuring of the low-energy spectrum, is demonstrated and matches the behavior predicted for quantum spin rings experiencing frustration between FM and AFM interactions.
Theoretical and Practical Implications
This work establishes several key insights for the molecular nanomagnetism domain:
- Essential role of non-Heisenberg terms: Both strong biquadratic exchange and DM components must be included to accurately model low-energy excitations, particularly as ring curvature and open 3d-shell occupancies increase.
- Curvature-driven DM interactions: Even in the absence of heavy elements or explicit SOC, ring geometry induces substantial DM-like exchange which meaningfully alters magnetic response.
- Electronic origins for nonzero DM in VM3+5: The anomalous DM terms in the VM3+6 chain point to a mechanism—potentially linked to the less-than-half-filled M3+7 subshell of VM3+8—requiring further many-body and current-density functional studies.
- Inter-site correlation criticality: DFT+M3+9+U0 functionals markedly improve the quantitative match with experiment, highlighting the necessity of accurate correlated electron treatments for reliable predictions.
From a practical standpoint, these findings impact the rational design of quantum magnetic materials for information storage, spintronics, and quantum coherence applications, where the correct hierarchy of competing interactions determines coherence times, excitation spectra, and operational temperature windows.
Future Directions
Immediate research avenues include explicit assessment of SOC, current-induced DM and biquadratic mechanisms, and the role of correlated spin-currents and non-collinear exchange-correlation fields beyond traditional functional constructions. Time-dependent DFT with Hubbard corrections may enable direct magnon spectrum prediction. The impact of chemical substitution, mechanical deformation, and symmetry breaking on these competing couplings merits investigation for targeted functionalization of molecular quantum devices.
Conclusion
A rigorous non-collinear ab initio treatment of CrU1 and VU2 molecular rings demonstrates that extended spin Hamiltonians—including biquadratic and DM-type couplings—are indispensable for quantitative description of spectral and magnetic properties. The research clarifies the interplay between geometry, electronic filling, and magnetism and sets a methodological benchmark for both fundamental exploration and applied quantum materials design.